

The Genetic and Epigenetic Footprint in Idiopathic Pulmonary Fibrosis and Familial Pulmonary Fibrosis: A State-of-the-Art Review
- PMID: 36553114
- PMCID: PMC9777399
- DOI: 10.3390/diagnostics12123107
The Genetic and Epigenetic Footprint in Idiopathic Pulmonary Fibrosis and Familial Pulmonary Fibrosis: A State-of-the-Art Review
Abstract
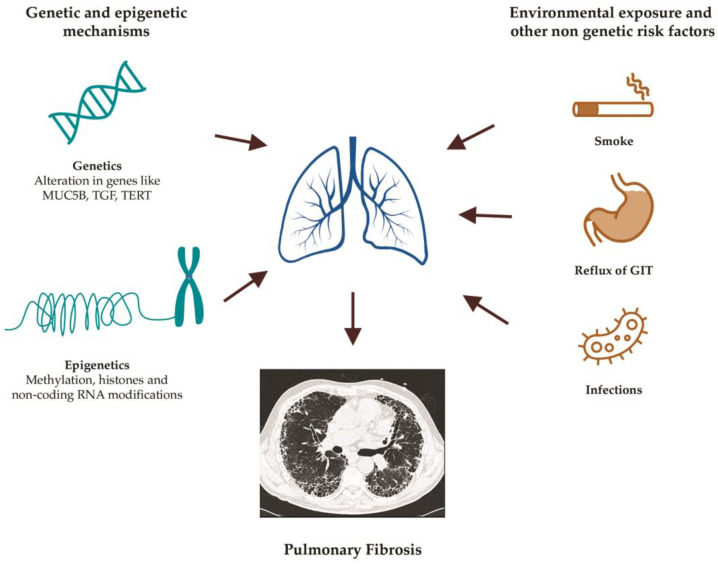
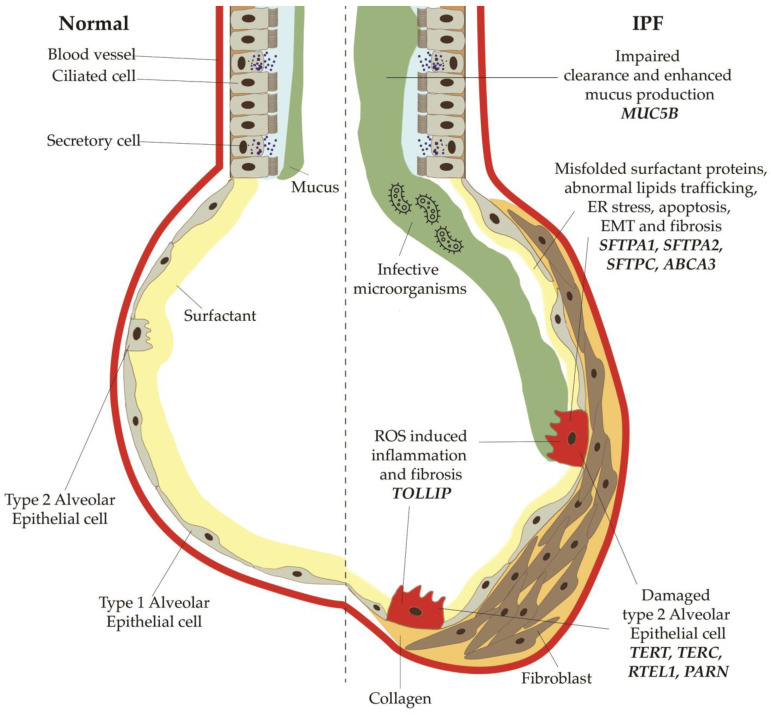
Idiopathic pulmonary fibrosis (IPF) is a rare disease of the lung with a largely unknown etiology and a poor prognosis. Intriguingly, forms of familial pulmonary fibrosis (FPF) have long been known and linked to specific genetic mutations. There is little evidence of the possible role of genetics in the etiology of sporadic IPF. We carried out a non-systematic, narrative literature review aimed at describing the main known genetic and epigenetic mechanisms that are involved in the pathogenesis and prognosis of IPF and FPF. In this review, we highlighted the mutations in classical genes associated with FPF, including those encoding for telomerases (TERT, TERC, PARN, RTEL1), which are also found in about 10-20% of cases of sporadic IPF. In addition to the Mendelian forms, mutations in the genes encoding for the surfactant proteins (SFTPC, SFTPA1, SFTPA2, ABCA3) and polymorphisms of genes for the mucin MUC5B and the Toll-interacting protein TOLLIP are other pathways favoring the fibrogenesis that have been thoroughly explored. Moreover, great attention has been paid to the main epigenetic alterations (DNA methylation, histone modification and non-coding RNA gene silencing) that are emerging to play a role in fibrogenesis. Finally, a gaze on the shared mechanisms between cancer and fibrogenesis, and future perspectives on the genetics of pulmonary fibrosis have been analyzed.
Keywords: ABCA3; MUC5B; epigenetic; familial pulmonary fibrosis; genetic; idiopathic pulmonary fibrosis; surfactant; telomerase; telomere; therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Raghu G., Remy-Jardin M., Myers J.L., Richeldi L., Ryerson C.J., Lederer D.J., Behr J., Cottin V., Danoff S.K., Morell F., et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. - DOI - PubMed
-
- Raghu G., Remy-Jardin M., Richeldi L., Thomson C.C., Inoue Y., Johkoh T., Kreuter M., Lynch D.A., Maher T.M., Martinez F.J., et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022;205:e18–e47. doi: 10.1164/rccm.202202-0399ST. - DOI - PMC - PubMed
-
- Tirelli C., Morandi V., Valentini A., La Carrubba C., Dore R., Zanframundo G., Morbini P., Grignaschi S., Franconeri A., Oggionni T., et al. Multidisciplinary Approach in the Early Detection of Undiagnosed Connective Tissue Diseases in Patients With Interstitial Lung Disease: A Retrospective Cohort Study. Front. Med. 2020;7:11. doi: 10.3389/fmed.2020.00011. - DOI - PMC - PubMed
-
- King T.E., Jr., Bradford W.Z., Castro-Bernardini S., Fagan E.A., Glaspole I., Glassberg M.K., Gorina E., Hopkins P.M., Kardatzke D., Lancaster L., et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
