

Previously Undescribed Gross HACE1 Deletions as a Cause of Autosomal Recessive Spastic Paraplegia
- PMID: 36553453
- PMCID: PMC9778407
- DOI: 10.3390/genes13122186
Previously Undescribed Gross HACE1 Deletions as a Cause of Autosomal Recessive Spastic Paraplegia
Abstract
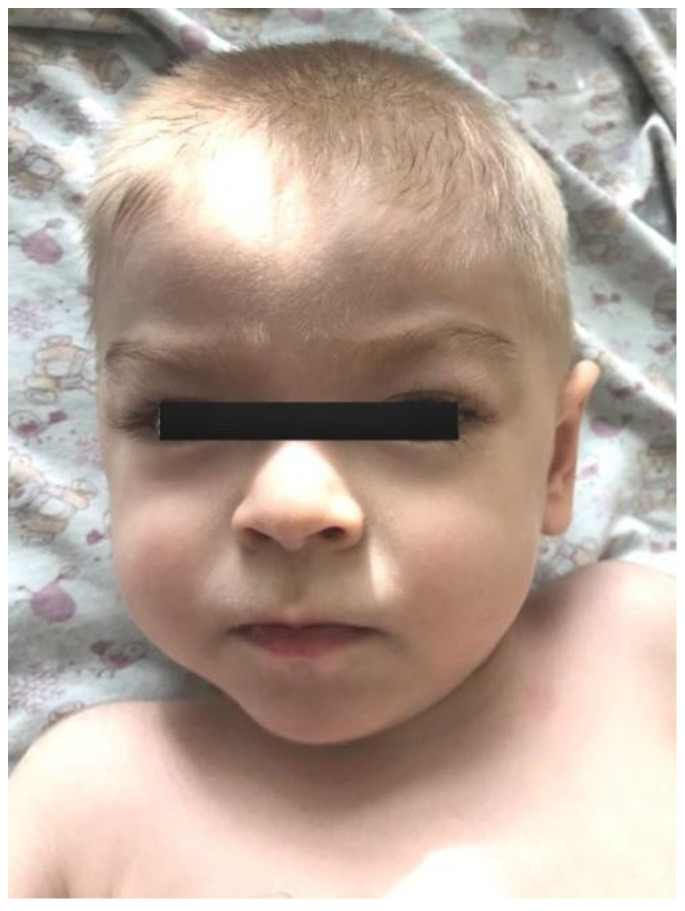
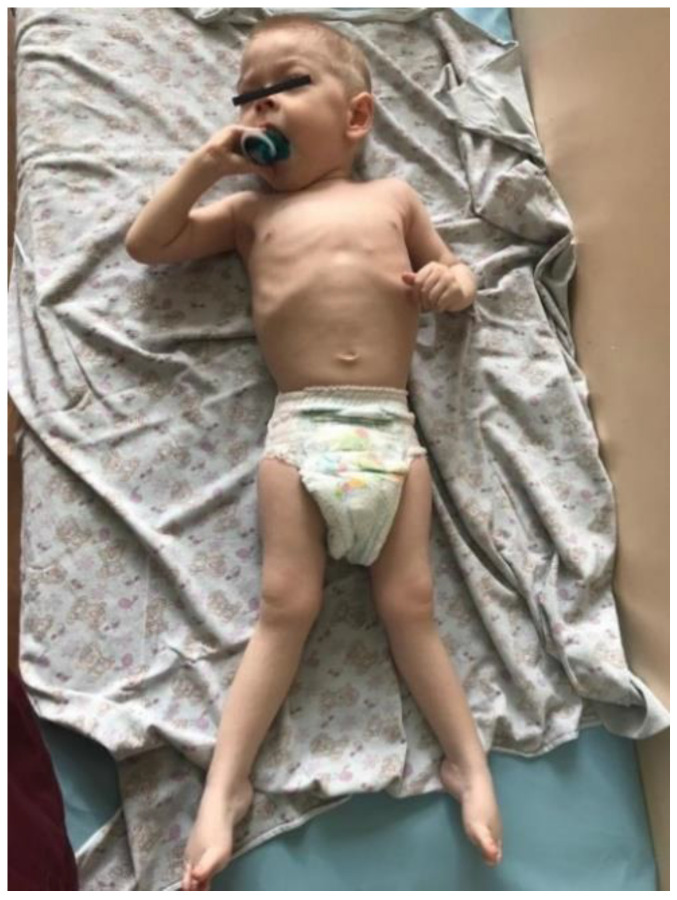
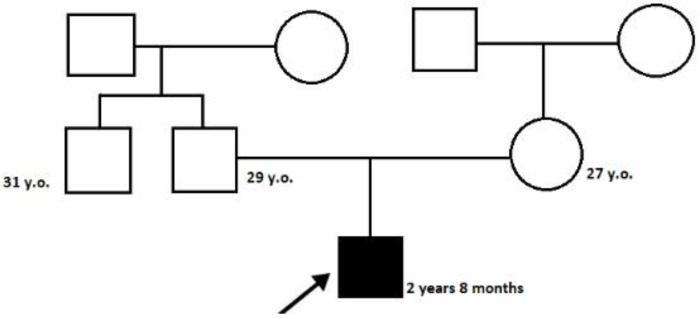
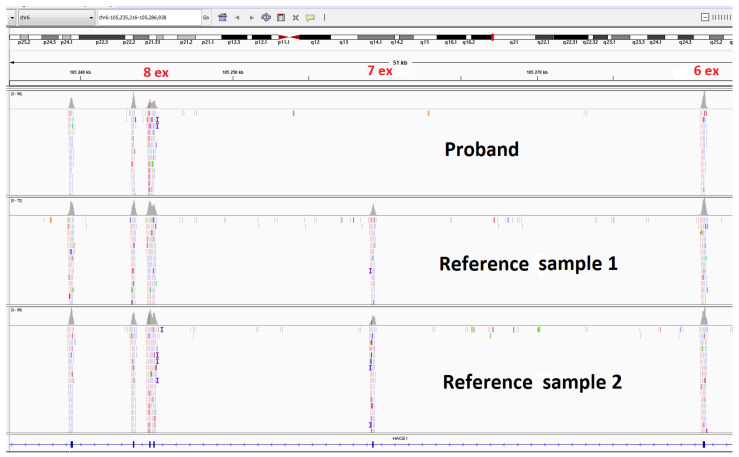
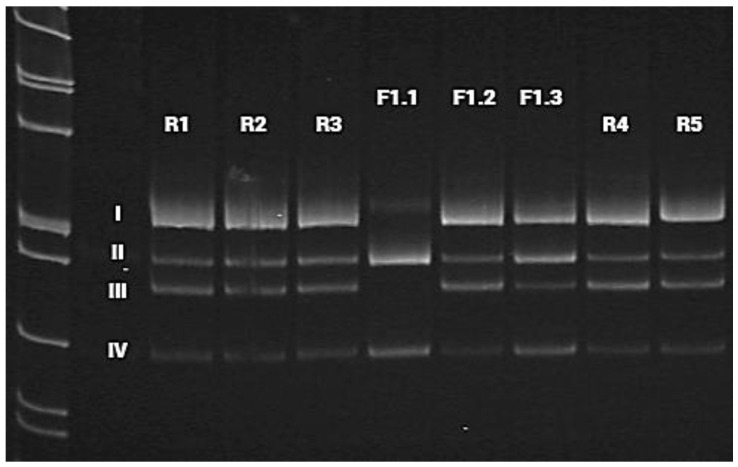
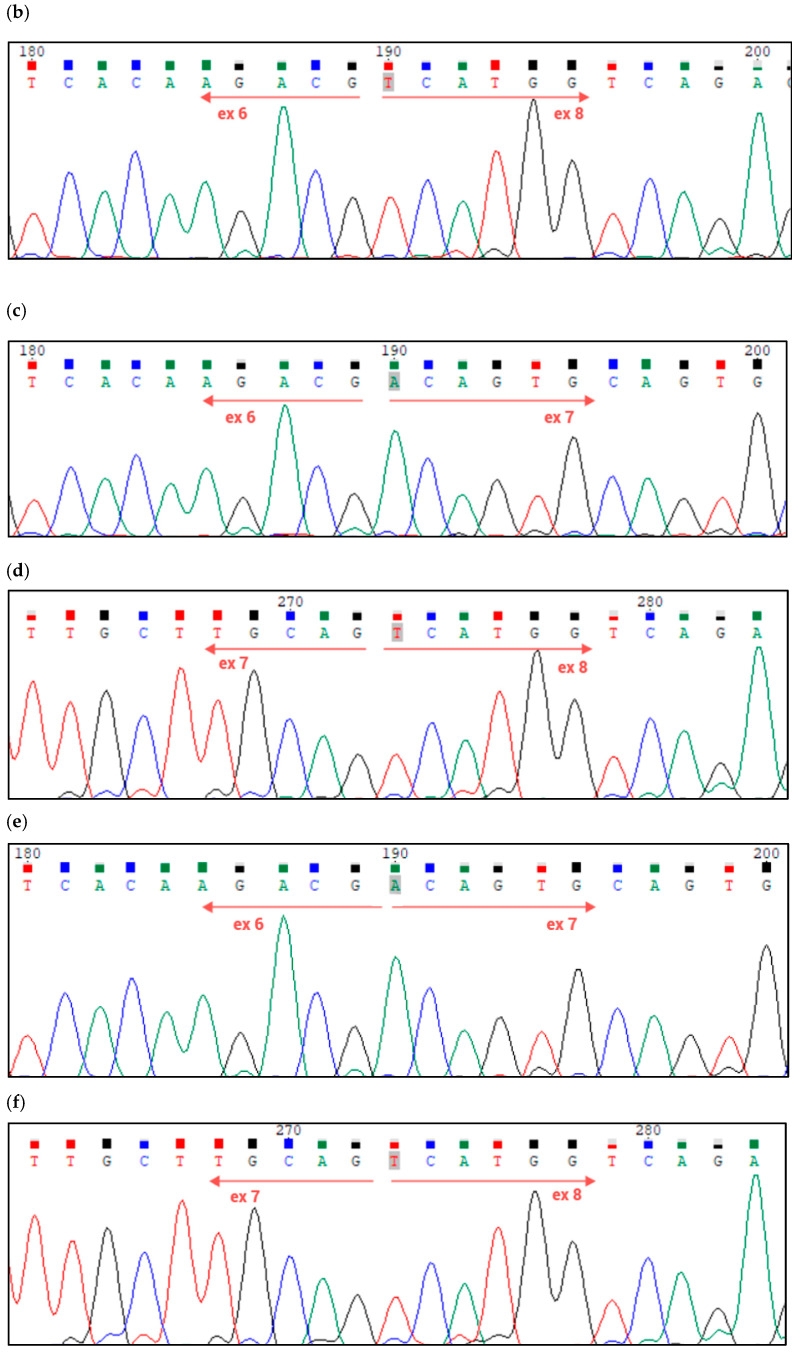
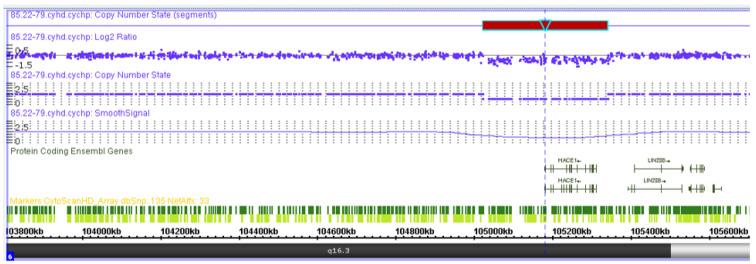
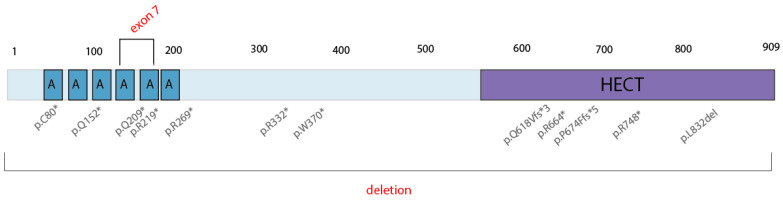
Spastic paraplegia and psychomotor retardation with or without seizures (SPPRS, OMIM 616756) is a rare genetic disease caused by biallelic pathogenic variants in the HACE1 gene. Originally, these mutations have been reported to be implicated in tumor predisposition. Nonetheless, via whole exome sequencing in 2015, HACE1 mutations were suggested to be the cause of a new autosomal recessive neurodevelopmental disorder, which is characterized by spasticity, muscular hypotonia, and intellectual disability. To date, 14 HACE1 pathogenic variants have been described; these variants have a loss-of-function effect that leads to clinical presentations with variable severities. However, gross deletions in the HACE1 gene have not yet been mentioned as a cause of spastic paraplegia. Here, we report a clinical case involving a 2-year-old male presenting with spasticity, mainly affecting the lower limbs, and developmental delay. Exome sequencing, chromosomal microarray analysis, and mRNA analysis were used to identify the causative gene. We revealed that the clinical findings were due to previously undescribed HACE1 biallelic deletions. We identified the deletion of exon 7: c.(534+1_535-1)_(617+1_618-1)del (NM_020771.4) and the gross deletion in the 6q16.3 locus, which affected the entire HACE1 gene: g.105018931_105337494del, (GRCh37). A comprehensive diagnostic approach for the patients with originally homozygous mutations in HACE1 is required since false homozygosity results are possible. More than 80% of the described mutations were reported to be homozygous. Initial hemizygosity is hard to detect by quantitative methods, and this may challenge molecular diagnostic identification in patients with spastic paraplegia.
Keywords: HACE1; neurodevelopmental syndrome; spastic paraplegia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
Autosomal Recessive Spastic Paraplegia and Psychomotor Retardation With or Without Seizures: A Case Report From Saudi Arabia.Cureus. 2024 May 20;16(5):e60642. doi: 10.7759/cureus.60642. eCollection 2024 May. Cureus. 2024. PMID: 38899231 Free PMC article.
-
[Clinical and genetic analysis of a child with Spastic paraplegia and psychomotor retardation with or without seizures due to compound heterozygous variants of the HACE1 gene].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025 Feb 10;42(2):156-161. doi: 10.3760/cma.j.cn511374-20240508-00278. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025. PMID: 40350393 Chinese.
-
A Retrospective Review of 18 Patients With Childhood-Onset Hereditary Spastic Paraplegia, Nine With Novel Variants.Pediatr Neurol. 2024 Mar;152:189-195. doi: 10.1016/j.pediatrneurol.2024.01.005. Epub 2024 Jan 6. Pediatr Neurol. 2024. PMID: 38301322
-
A novel homozygous variant in RNF170 causes hereditary spastic paraplegia: a case report and review of the literature.Neurogenetics. 2022 Apr;23(2):85-90. doi: 10.1007/s10048-022-00685-6. Epub 2022 Jan 18. Neurogenetics. 2022. PMID: 35041108 Review.
-
A novel homozygous mutation in ERLIN1 gene causing spastic paraplegia 62 and literature review.Eur J Med Genet. 2022 Nov;65(11):104608. doi: 10.1016/j.ejmg.2022.104608. Epub 2022 Sep 12. Eur J Med Genet. 2022. PMID: 36100157 Review.
Cited by
-
Autosomal Recessive Spastic Paraplegia and Psychomotor Retardation With or Without Seizures: A Case Report From Saudi Arabia.Cureus. 2024 May 20;16(5):e60642. doi: 10.7759/cureus.60642. eCollection 2024 May. Cureus. 2024. PMID: 38899231 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
