

Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis
- PMID: 36556960
- PMCID: PMC9784142
- DOI: 10.3390/medicina58121757
Clinical and Radiological Features of Interstitial Lung Diseases Associated with Polymyositis and Dermatomyositis
Abstract
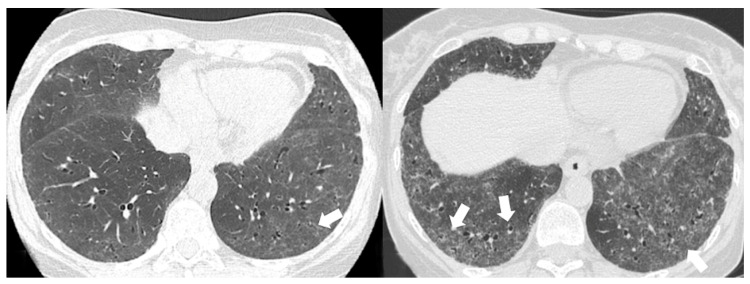
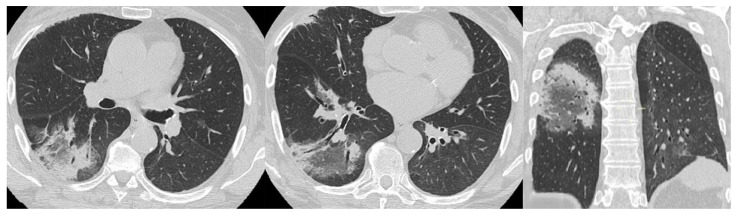
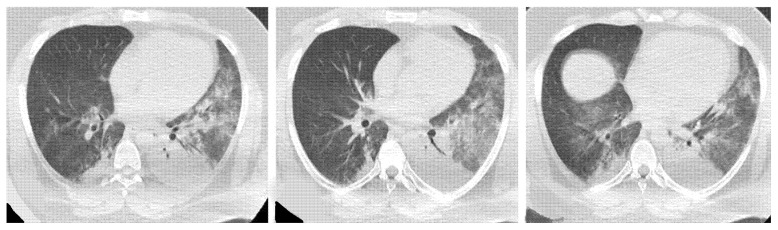
Polymyositis and dermatomyositis are autoimmune idiopathic systemic inflammatory diseases, characterized by various degrees of muscle inflammation and typical cutaneous lesions-the latter found in dermatomyositis. The underlying pathogenesis is characterized by a high level of uncertainty, and recent studies suggest diseases may have different immunopathological mechanisms. In polymyositis, components of the cellular immune system are involved, whereas in dermatomyositis, the pathogenesis is mainly mediated by the humoral immune response. The interstitial lung disease occurs in one-third of polymyositis and dermatomyositis patients associated with worse outcomes, showing an estimated excess mortality rate of around 40%. Lung involvement may also appear, such as a complication of muscle weakness, mainly represented by aspiration pneumonia or respiratory insufficiency. The clinical picture is characterized, in most cases, by progressive dyspnea and non-productive cough. In some cases, hemoptysis and chest pain are found. Onset can be acute, sub-acute, or chronic. Pulmonary involvement could be assessed by High Resolution Computed Tomography (HRCT), which may identify early manifestations of diseases. Moreover, Computed Tomography (CT) appearances can be highly variable depending on the positivity of myositis-specific autoantibodies. The most common pathological patterns include fibrotic and cellular nonspecific interstitial pneumonia or organizing pneumonia; major findings observed on HRCT images are represented by consolidations, ground-glass opacities, and reticulations. Other findings include honeycombing, subpleural bands, and traction bronchiectasis. In patients having Anti-ARS Abs, HRCT features may develop with consolidations, ground glass opacities (GGOs), and reticular opacities in the peripheral portions; nonspecific interstitial pneumonia or nonspecific interstitial pneumonia mixed with organizing pneumonia have been reported as the most frequently encountered patterns. In patients with anti-MDA5 Abs, mixed or unclassifiable patterns are frequently observed at imaging. HRCT is a sensitive method that allows one not only to identify disease, but also to monitor the effectiveness of treatment and detect disease progression and/or complications; however, radiological findings are not specific. Therefore, aim of this pictorial essay is to describe clinical and radiological features of interstitial lung diseases associated with polymyositis and dermatomyositis, emphasizing the concept that gold standard for diagnosis and classification-should be based on a multidisciplinary approach.
Keywords: autoimmune diseases; dermatomyositis; lung disease interstitial; multidetector computed tomography; polymyositis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures











References
-
- Hayashi S., Tanaka M., Kobayashi H., Nakazono T., Satoh T., Fukuno Y., Aragane N., Tada Y., Koarada S., Ohta A., et al. High-resolution computed tomography characterization of interstitial lung diseases in polymyositis/dermatomyositis. J. Rheumatol. 2008;35:260–269. - PubMed
-
- Sugiyama Y., Yoshimi R., Tamura M., Takeno M., Kunishita Y., Kishimoto D., Yoshioka Y., Kobayashi K., Takase-Minegishi K., Watanabe T., et al. The predictive prognostic factors for polymyositis/dermatomyositis-associated interstitial lung disease. Arthritis Res. Ther. 2018;20:7. doi: 10.1186/s13075-017-1506-7. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
