

Statins Protect Against Early Stages of Doxorubicin-induced Cardiotoxicity Through the Regulation of Akt Signaling and SERCA2
- PMID: 36562012
- PMCID: PMC9764135
- DOI: 10.1016/j.cjco.2022.08.006
Statins Protect Against Early Stages of Doxorubicin-induced Cardiotoxicity Through the Regulation of Akt Signaling and SERCA2
Abstract
Background: Doxorubicin-induced cardiomyopathy (DICM) is one of the complications that can limit treatment for a significant number of cancer patients. In animal models, the administration of statins can prevent the development of DICM. Therefore, the use of statins with anthracyclines potentially could enable cancer patients to complete their chemotherapy without added cardiotoxicity. The precise mechanism mediating the cardioprotection is not well understood. The purpose of this study is to determine the molecular mechanism by which rosuvastatin confers cardioprotection in a mouse model of DICM.
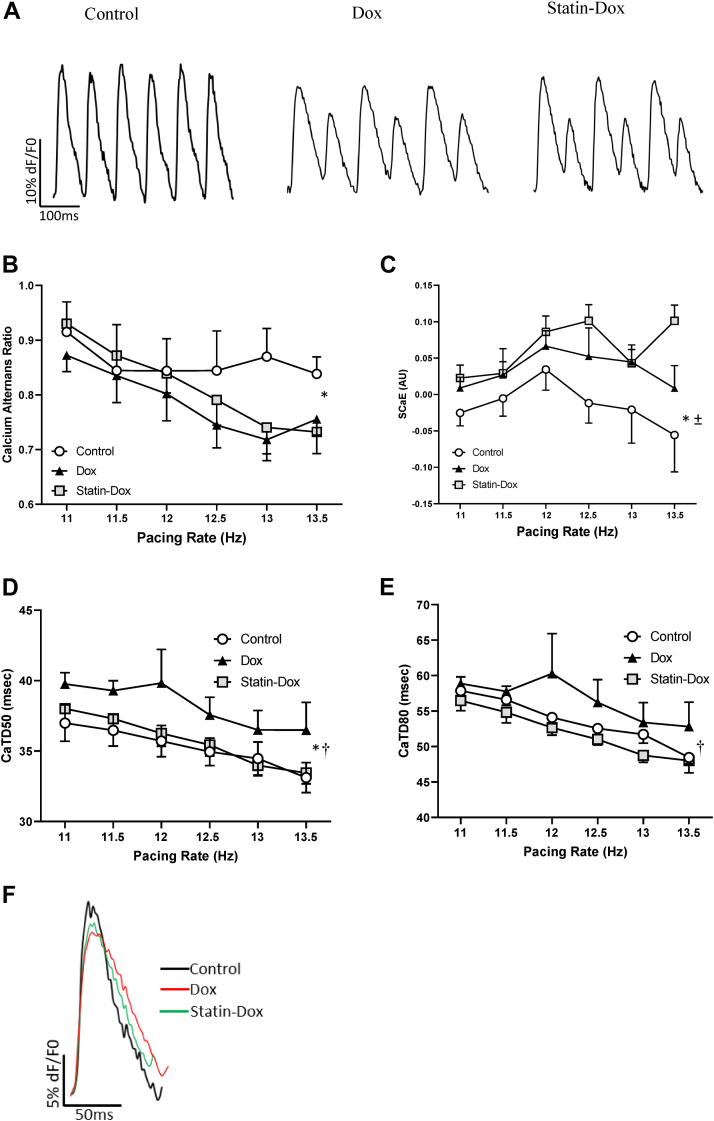
Methods: Rosuvastatin was intraperitoneally administered into adult male mice at 100 μg/kg daily for 7 days, followed by a single intraperitoneal doxorubicin injection at 10 mg/kg. Animals continued to receive rosuvastatin daily for an additional 14 days. Cardiac function was assessed by echocardiography. Optical calcium mapping was performed on retrograde Langendorff perfused isolated hearts. Ventricular tissue samples were analyzed by immunofluorescence microscopy, Western blotting, and quantitative polymerase chain reaction.
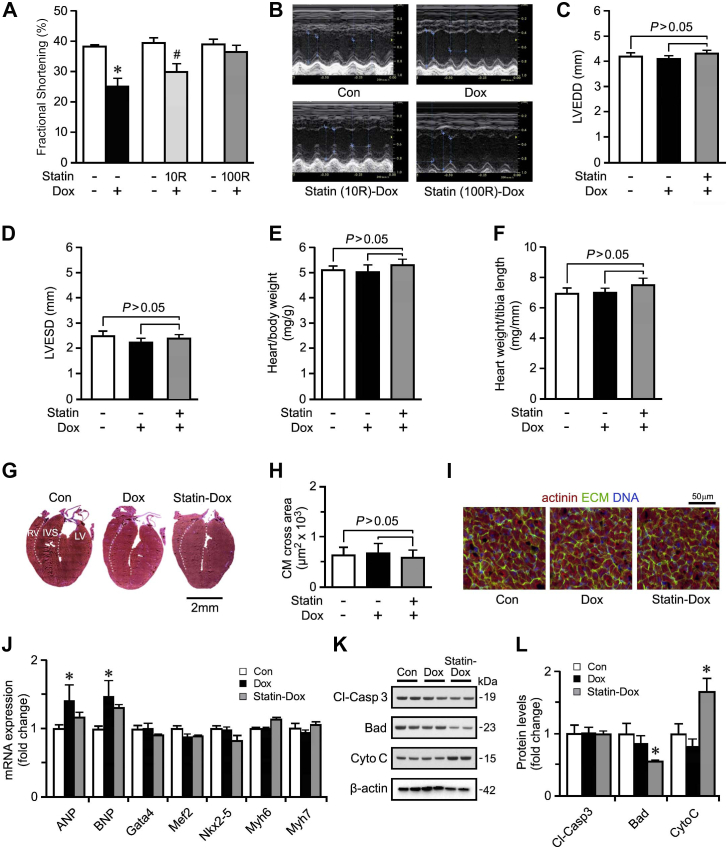
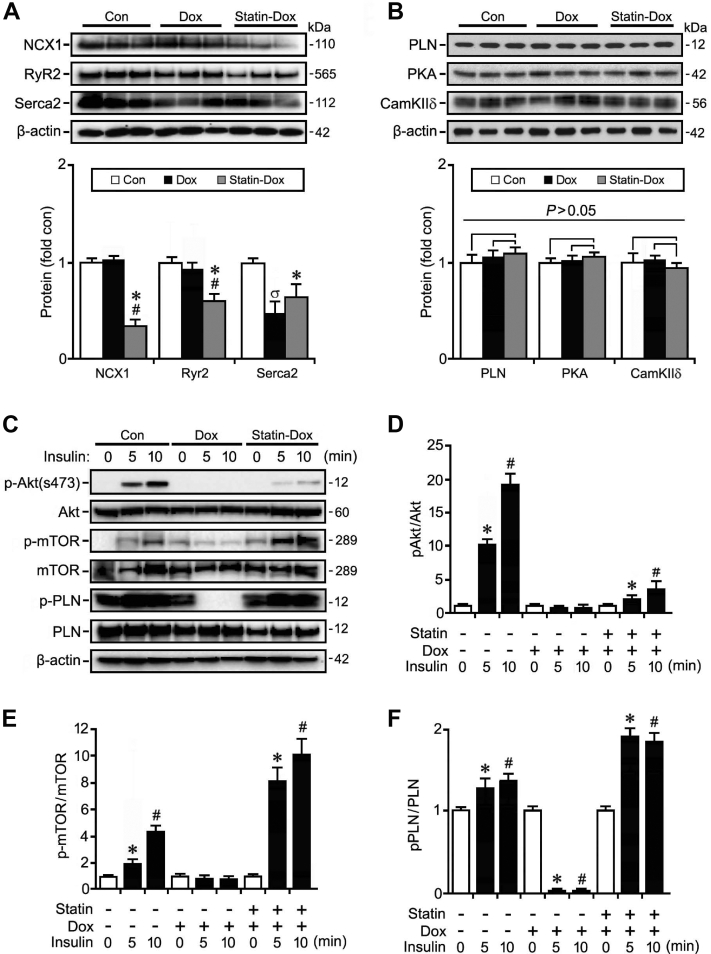
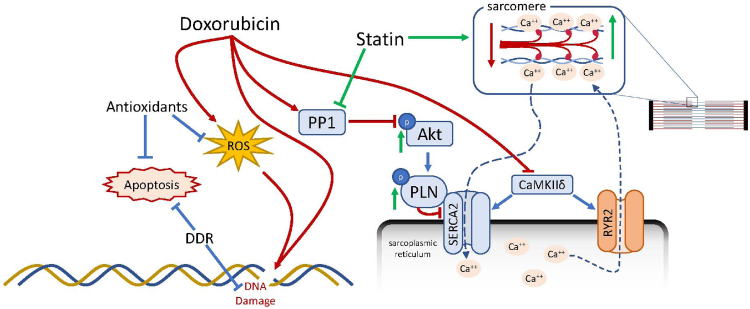
Results: Exposure to doxorubicin resulted in significantly reduced fractional shortening (27.4% ± 1.11% vs 40% ± 5.8% in controls; P < 0.001) and re-expression of the fetal gene program. However, we found no evidence of maladaptive cardiac hypertrophy or adverse ventricular remodeling in mice exposed to this dose of doxorubicin. In contrast, rosuvastatin-doxorubicin-treated mice maintained their cardiac function (39% ± 1.26%; P < 0.001). Mechanistically, the effect of rosuvastatin was associated with activation of Akt and phosphorylation of phospholamban with preserved sarcoplasmic/endoplasmic reticulum Ca2+ transporting 2 (SERCA2)-mediated Ca2+ reuptake. These effects occurred independently of perturbations in ryanodine receptor 2 function.
Conclusions: Rosuvastatin counteracts the cardiotoxic effects of doxorubicin by directly targeting sarcoplasmic calcium cycling.
Contexte: La cardiomyopathie induite par la doxorubicine (CMID) est l’une des complications pouvant limiter le traitement d’un nombre considérable de patients atteints de cancer. Dans des modèles animaux, l’administration de statines peut prévenir l’apparition d’une CMID. Ainsi, l’utilisation de statines avec les anthracyclines pourrait vraisemblablement permettre aux patients de compléter leur chimiothérapie en évitant une cardiotoxicité supplémentaire. Le mécanisme précis qui sous-tend cet effet cardioprotecteur n’est pas entièrement élucidé. Cette étude a pour objectif de déterminer dans un modèle murin de CMID le mécanisme moléculaire par lequel la rosuvastatine confère une cardioprotection.
Méthodologie: La rosuvastatine a été administrée par voie intrapéritonéale à des souris adultes mâles à une dose de 100 μg/kg par jour pendant sept jours, suivie d’une dose unique de doxorubicine de 10 mg/kg administrée par injection intrapéritonéale. Les animaux poursuivaient ensuite le traitement par la rosuvastatine une fois par jour pendant 14 jours supplémentaires. La fonction cardiaque a été mesurée par échocardiographie. Une cartographie optique du calcium a été réalisée sur des cœurs isolés soumis à une perfusion rétrograde selon la méthode de Langendorff. Des échantillons de tissu ventriculaire ont été analysés par microscopie en immunofluorescence, par buvardage de western et par mesure quantitative de l’amplification en chaîne par polymérase.
Résultats: L’exposition à la doxorubicine a entraîné une diminution significative de la fraction de raccourcissement (27,4 % ± 1,11 % vs 40 % ± 5,8 % dans le groupe témoin; p < 0,001) et la réexpression du programme génique fœtal. Toutefois, aucune hypertrophie cardiaque inadaptée ni aucun remodelage ventriculaire indésirable n’ont été observés chez les souris ayant été exposées à la dose de doxorubicine étudiée. En revanche, la fonction cardiaque a été préservée chez les souris traitées par l’association rosuvastatine-doxorubicine (39 % ± 1,26 %; p < 0,001). Sur le plan du mode d’action, l’effet de la rosuvastatine a été associé à une activation de l’Akt et à une phosphorylation du phospholambane, avec préservation du recaptage de Ca2+ médié par la pompe SERCA2 (sarcoplasmic/endoplasmic reticulum Ca 2+ transporting 2). Ces effets sont survenus indépendamment des perturbations de la fonction du récepteur RyR2 (ryanodine receptor 2).
Conclusions: La rosuvastatine neutralise les effets cardiotoxiques de la doxorubicine en ciblant directement la circulation sarcoplasmique du calcium.
© 2022 The Authors.
Figures
Similar articles
-
Doxorubicin-induced delayed-onset subclinical cardiotoxicity in mice.J Appl Toxicol. 2022 May;42(5):778-792. doi: 10.1002/jat.4256. Epub 2021 Oct 20. J Appl Toxicol. 2022. PMID: 34668590
-
Acylated Ghrelin Protects the Hearts of Rats from Doxorubicin-Induced Fas/FasL Apoptosis by Stimulating SERCA2a Mediated by Activation of PKA and Akt.Cardiovasc Toxicol. 2019 Dec;19(6):529-547. doi: 10.1007/s12012-019-09527-8. Cardiovasc Toxicol. 2019. PMID: 31093930
-
Cardiotoxic Effects of Short-Term Doxorubicin Administration: Involvement of Connexin 43 in Calcium Impairment.Int J Mol Sci. 2017 Oct 11;18(10):2121. doi: 10.3390/ijms18102121. Int J Mol Sci. 2017. PMID: 29019935 Free PMC article.
-
S100A8 and S100A9 Are Associated with Doxorubicin-Induced Cardiotoxicity in the Heart of Diabetic Mice.Front Physiol. 2016 Aug 5;7:334. doi: 10.3389/fphys.2016.00334. eCollection 2016. Front Physiol. 2016. PMID: 27547188 Free PMC article.
-
Major vault protein attenuates cardiomyocyte injury in doxorubicin-induced cardiomyopathy through activating AKT.BMC Cardiovasc Disord. 2022 Mar 4;22(1):77. doi: 10.1186/s12872-022-02517-9. BMC Cardiovasc Disord. 2022. PMID: 35246039 Free PMC article.
Cited by
-
Statins to prevent early cardiac dysfunction in cancer patients at increased cardiotoxicity risk receiving anthracyclines.Eur Heart J Cardiovasc Pharmacother. 2023 Sep 20;9(6):515-525. doi: 10.1093/ehjcvp/pvad031. Eur Heart J Cardiovasc Pharmacother. 2023. PMID: 37120736 Free PMC article. Clinical Trial.
-
Statins: Novel Approaches for the Management of Doxorubicin-Induced Cardiotoxicity-A Literature Review.Cardiovasc Toxicol. 2025 Sep;25(9):1429-1452. doi: 10.1007/s12012-025-10030-6. Epub 2025 Jul 10. Cardiovasc Toxicol. 2025. PMID: 40637833 Review.
-
Recent Advances in the Mechanisms of Cell Death and Dysfunction in Doxorubicin Cardiotoxicity.Rev Cardiovasc Med. 2023 Nov 27;24(11):336. doi: 10.31083/j.rcm2411336. eCollection 2023 Nov. Rev Cardiovasc Med. 2023. PMID: 39076437 Free PMC article. Review.
-
Mitophagy in Doxorubicin-Induced Cardiotoxicity: Insights into Molecular Biology and Novel Therapeutic Strategies.Biomolecules. 2024 Dec 17;14(12):1614. doi: 10.3390/biom14121614. Biomolecules. 2024. PMID: 39766321 Free PMC article. Review.
-
Cardiomyopathies and a brief insight into DOX-induced cardiomyopathy.Egypt Heart J. 2025 Mar 10;77(1):29. doi: 10.1186/s43044-025-00628-0. Egypt Heart J. 2025. PMID: 40064787 Free PMC article. Review.
References
-
- Yoshida M., Shiojima I., Ikeda H., Komuro I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J Mol Cell Cardiol. 2009;47:698–705. - PubMed
-
- Myers C. The role of iron in doxorubicin-induced cardiomyopathy. Semin Oncol. 1998;25:10–14. - PubMed
-
- Octavia Y., Tocchetti C.G., Gabrielson K.L., et al. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012;52:1213–1225. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
