

Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits
- PMID: 36572924
- PMCID: PMC9793634
- DOI: 10.1186/s40168-022-01424-7
Global patterns of diversity and metabolism of microbial communities in deep-sea hydrothermal vent deposits
Abstract
Background: When deep-sea hydrothermal fluids mix with cold oxygenated fluids, minerals precipitate out of solution and form hydrothermal deposits. These actively venting deep-sea hydrothermal deposits support a rich diversity of thermophilic microorganisms which are involved in a range of carbon, sulfur, nitrogen, and hydrogen metabolisms. Global patterns of thermophilic microbial diversity in deep-sea hydrothermal ecosystems have illustrated the strong connectivity between geological processes and microbial colonization, but little is known about the genomic diversity and physiological potential of these novel taxa. Here we explore this genomic diversity in 42 metagenomes from four deep-sea hydrothermal vent fields and a deep-sea volcano collected from 2004 to 2018 and document their potential implications in biogeochemical cycles.
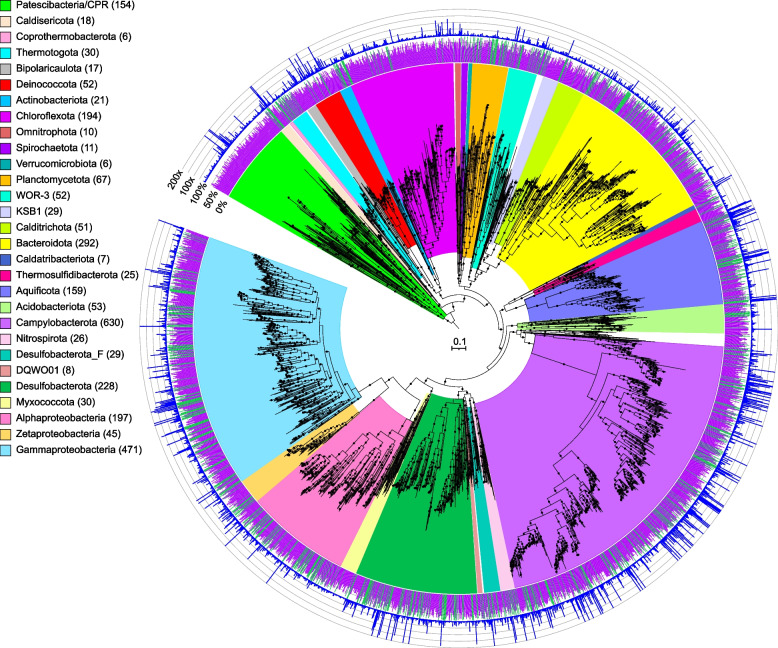
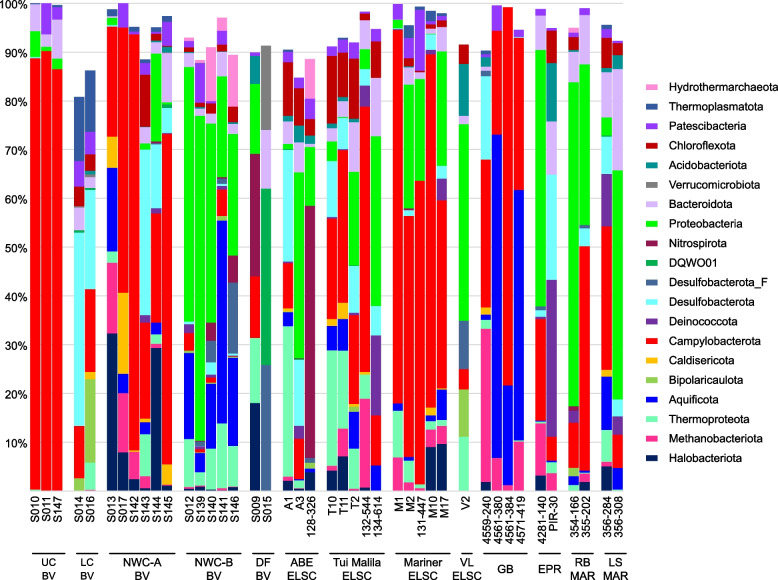
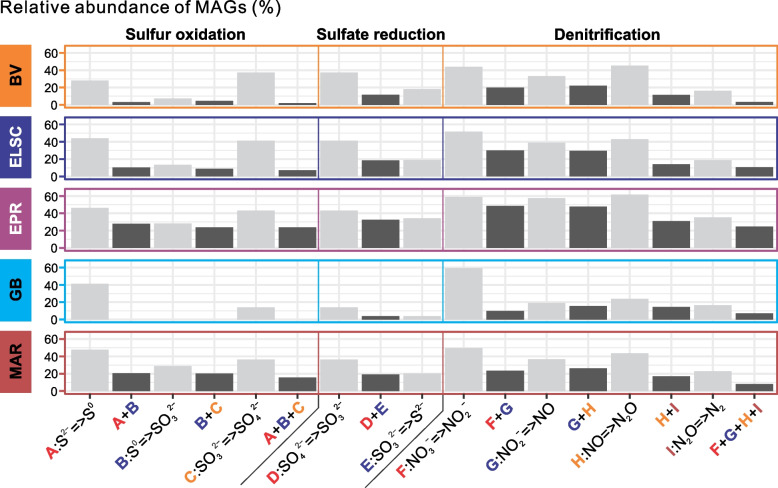
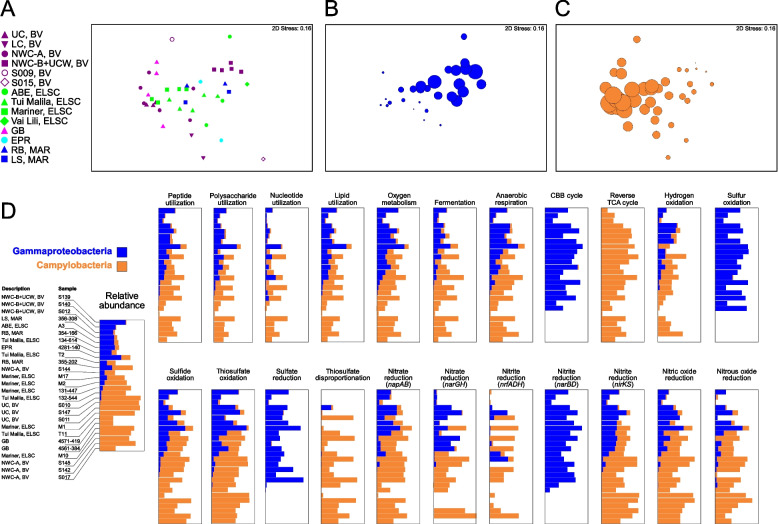
Results: Our dataset represents 3635 metagenome-assembled genomes encompassing 511 novel and recently identified genera from deep-sea hydrothermal settings. Some of the novel bacterial (107) and archaeal genera (30) that were recently reported from the deep-sea Brothers volcano were also detected at the deep-sea hydrothermal vent fields, while 99 bacterial and 54 archaeal genera were endemic to the deep-sea Brothers volcano deposits. We report some of the first examples of medium- (≥ 50% complete, ≤ 10% contaminated) to high-quality (> 90% complete, < 5% contaminated) MAGs from phyla and families never previously identified, or poorly sampled, from deep-sea hydrothermal environments. We greatly expand the novel diversity of Thermoproteia, Patescibacteria (Candidate Phyla Radiation, CPR), and Chloroflexota found at deep-sea hydrothermal vents and identify a small sampling of two potentially novel phyla, designated JALSQH01 and JALWCF01. Metabolic pathway analysis of metagenomes provides insights into the prevalent carbon, nitrogen, sulfur, and hydrogen metabolic processes across all sites and illustrates sulfur and nitrogen metabolic "handoffs" in community interactions. We confirm that Campylobacteria and Gammaproteobacteria occupy similar ecological guilds but their prevalence in a particular site is driven by shifts in the geochemical environment.
Conclusion: Our study of globally distributed hydrothermal vent deposits provides a significant expansion of microbial genomic diversity associated with hydrothermal vent deposits and highlights the metabolic adaptation of taxonomic guilds. Collectively, our results illustrate the importance of comparative biodiversity studies in establishing patterns of shared phylogenetic diversity and physiological ecology, while providing many targets for enrichment and cultivation of novel and endemic taxa. Video Abstract.
Keywords: Archaea; Bacteria; Deep-sea hydrothermal vents; Metagenomics; Thermophiles.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Nakagawa S, Takai K, Inagaki F, Chiba H, Ishibashi JI, Kataoka S, et al. Variability in microbial community and venting chemistry in a sediment-hosted backarc hydrothermal system: impacts of subseafloor phase-separation. FEMS Microbiol Ecol. 2005;54:141–155. doi: 10.1016/j.femsec.2005.03.007. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
