

TBK1 phosphorylation activates LIR-dependent degradation of the inflammation repressor TNIP1
- PMID: 36574265
- PMCID: PMC9797988
- DOI: 10.1083/jcb.202108144
TBK1 phosphorylation activates LIR-dependent degradation of the inflammation repressor TNIP1
Abstract
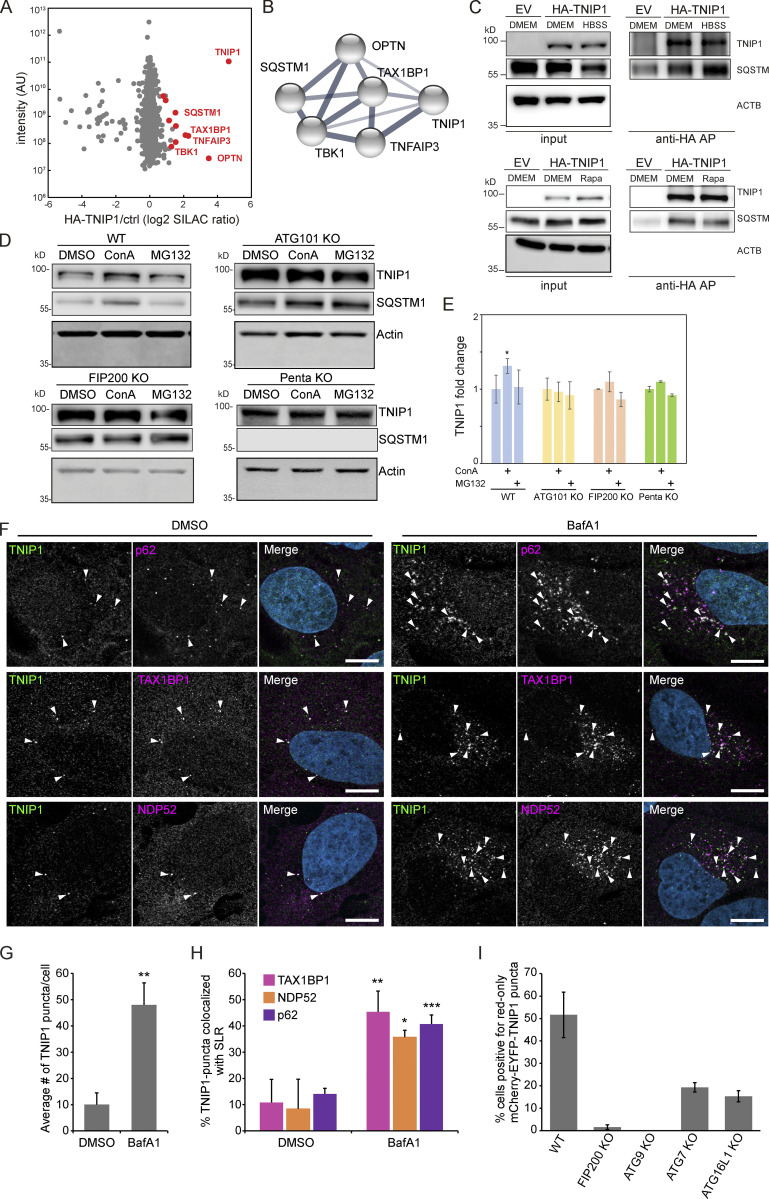
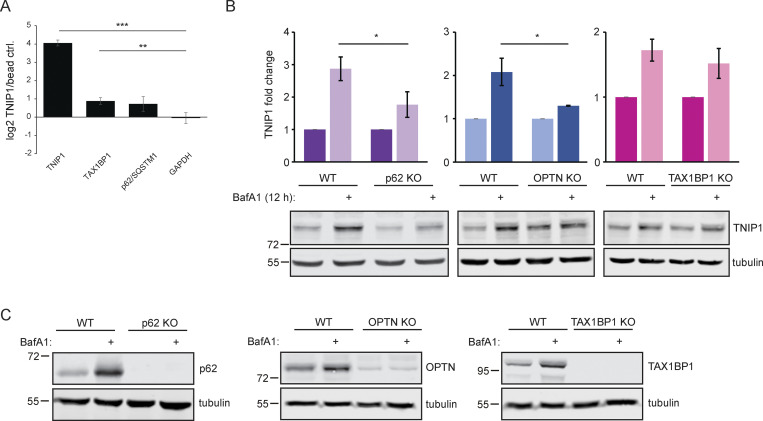
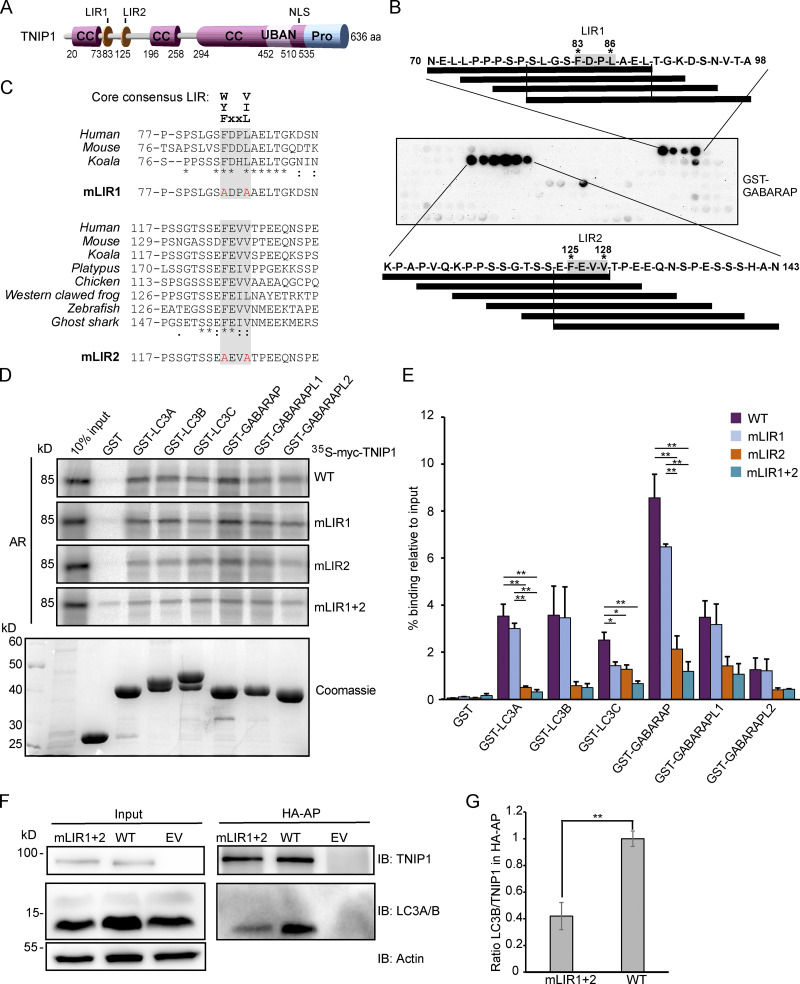
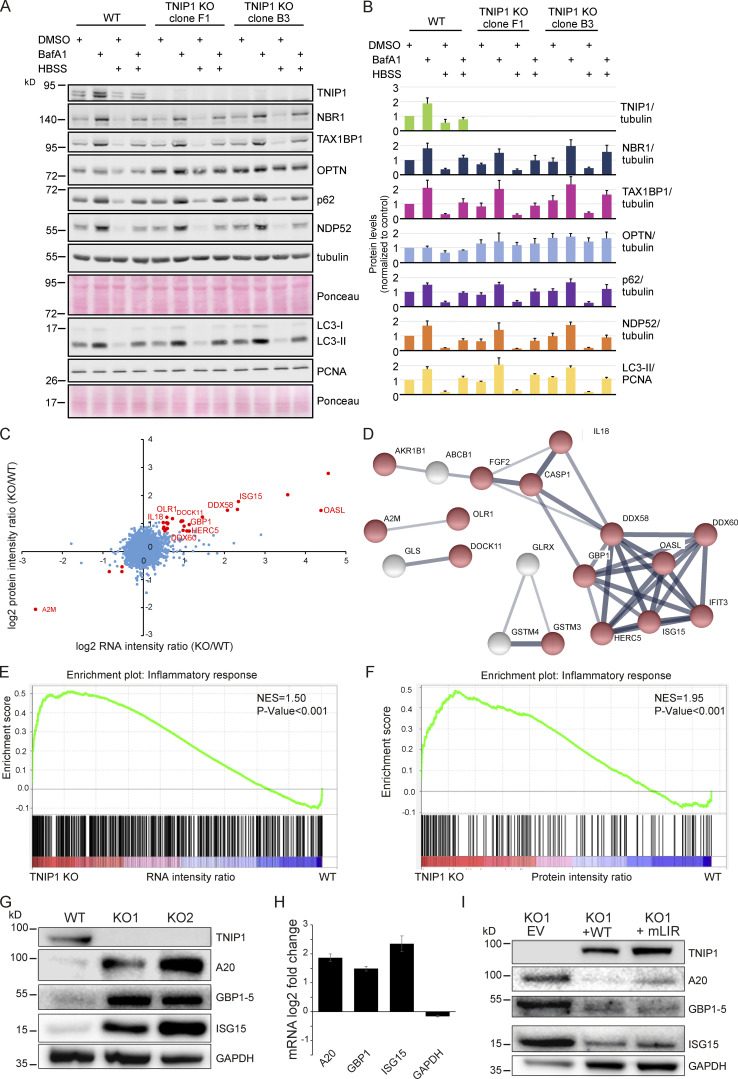
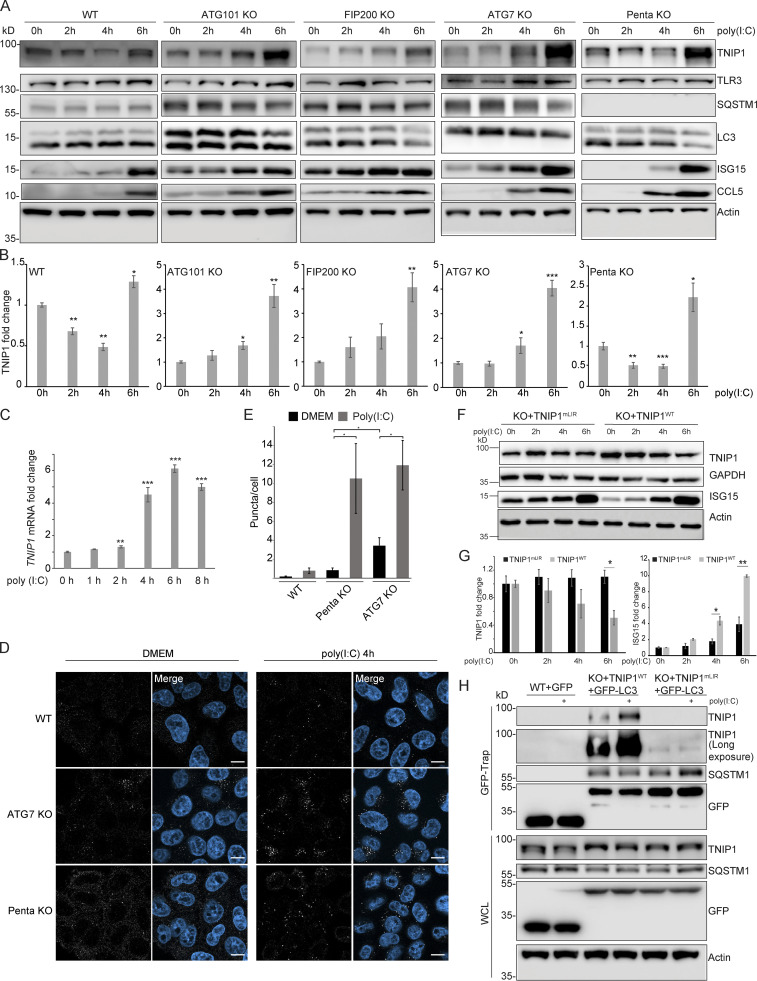
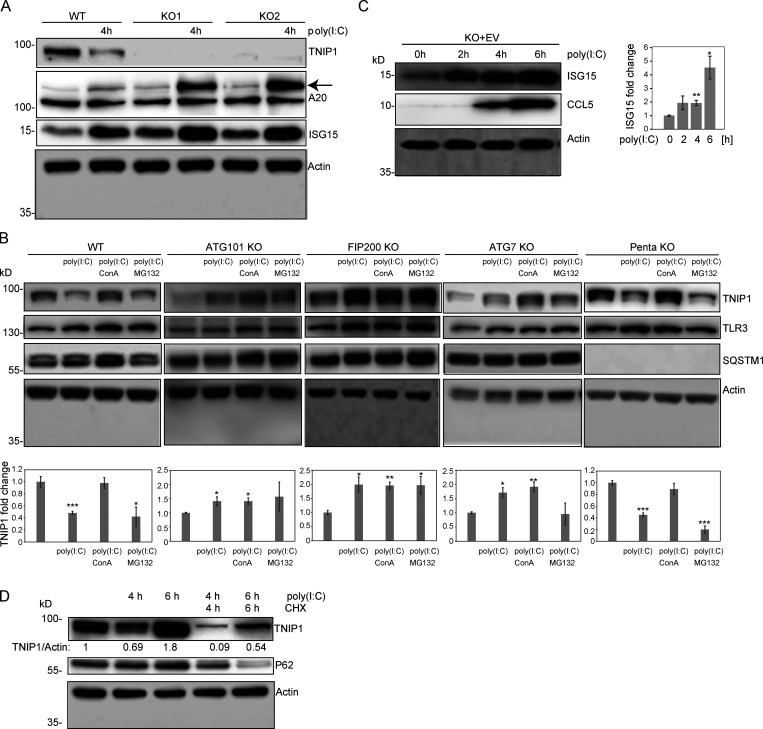
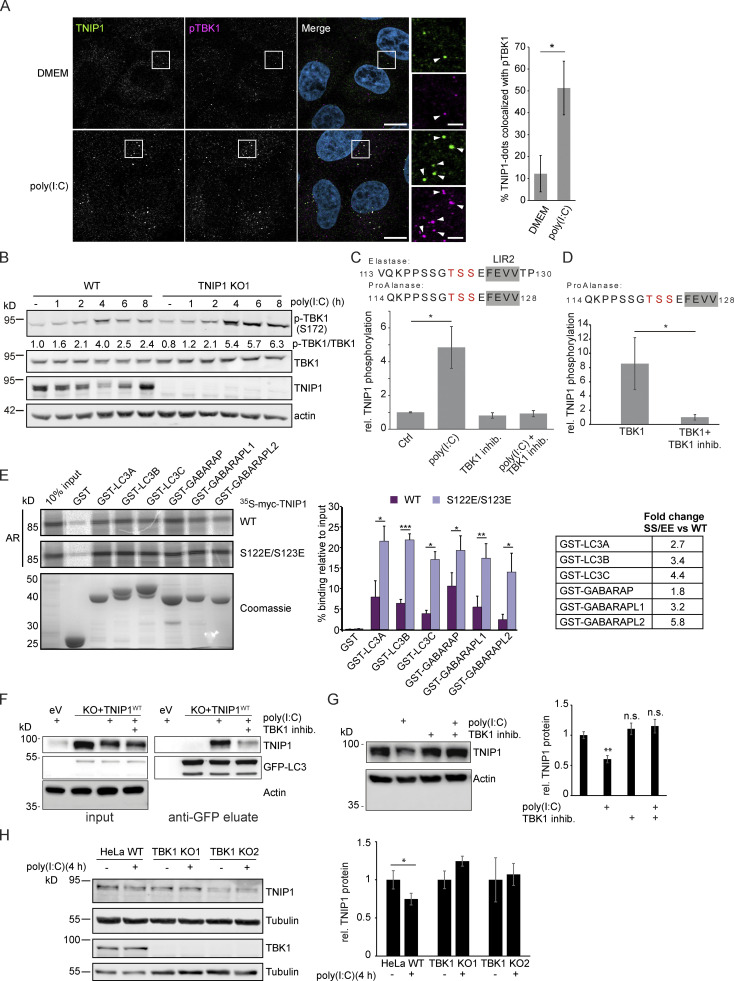
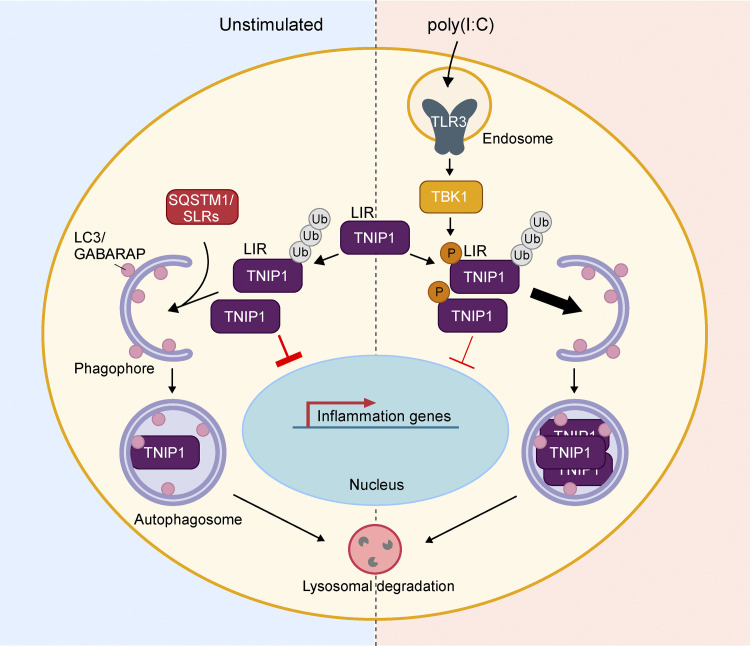
Limitation of excessive inflammation due to selective degradation of pro-inflammatory proteins is one of the cytoprotective functions attributed to autophagy. In the current study, we highlight that selective autophagy also plays a vital role in promoting the establishment of a robust inflammatory response. Under inflammatory conditions, here TLR3-activation by poly(I:C) treatment, the inflammation repressor TNIP1 (TNFAIP3 interacting protein 1) is phosphorylated by Tank-binding kinase 1 (TBK1) activating an LIR motif that leads to the selective autophagy-dependent degradation of TNIP1, supporting the expression of pro-inflammatory genes and proteins. This selective autophagy efficiently reduces TNIP1 protein levels early (0-4 h) upon poly(I:C) treatment to allow efficient initiation of the inflammatory response. At 6 h, TNIP1 levels are restored due to increased transcription avoiding sustained inflammation. Thus, similarly as in cancer, autophagy may play a dual role in controlling inflammation depending on the exact state and timing of the inflammatory response.
© 2022 Zhou et al.
Conflict of interest statement
Disclosures: The authors declare no competing financial interests.
Figures
Similar articles
-
The inflammation repressor TNIP1/ABIN-1 is degraded by autophagy following TBK1 phosphorylation of its LIR.Autophagy. 2023 Oct;19(10):2819-2820. doi: 10.1080/15548627.2023.2185013. Epub 2023 Mar 9. Autophagy. 2023. PMID: 36847414 Free PMC article.
-
TNIP1 inhibits selective autophagy via bipartite interaction with LC3/GABARAP and TAX1BP1.Mol Cell. 2023 Mar 16;83(6):927-941.e8. doi: 10.1016/j.molcel.2023.02.023. Epub 2023 Mar 9. Mol Cell. 2023. PMID: 36898370 Free PMC article.
-
NIMA-related kinase 9-mediated phosphorylation of the microtubule-associated LC3B protein at Thr-50 suppresses selective autophagy of p62/sequestosome 1.J Biol Chem. 2020 Jan 31;295(5):1240-1260. doi: 10.1074/jbc.RA119.010068. Epub 2019 Dec 19. J Biol Chem. 2020. PMID: 31857374 Free PMC article.
-
A review on the role of TANK-binding kinase 1 signaling in cancer.Int J Biol Macromol. 2021 Jul 31;183:2364-2375. doi: 10.1016/j.ijbiomac.2021.06.022. Epub 2021 Jun 7. Int J Biol Macromol. 2021. PMID: 34111484 Review.
-
Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles.Autophagy. 2021 Feb;17(2):385-401. doi: 10.1080/15548627.2020.1725377. Epub 2020 Feb 12. Autophagy. 2021. PMID: 32048886 Free PMC article. Review.
Cited by
-
In Silico Insights: QSAR Modeling of TBK1 Kinase Inhibitors for Enhanced Drug Discovery.J Chem Inf Model. 2024 Oct 14;64(19):7488-7502. doi: 10.1021/acs.jcim.4c00864. Epub 2024 Sep 17. J Chem Inf Model. 2024. PMID: 39289178 Free PMC article.
-
Autophagy receptor OsNBR1 modulates salt stress tolerance in rice.Plant Cell Rep. 2023 Dec 25;43(1):17. doi: 10.1007/s00299-023-03111-9. Plant Cell Rep. 2023. PMID: 38145426
-
Targeted proteomics addresses selectivity and complexity of protein degradation by autophagy.Autophagy. 2025 Feb;21(2):460-475. doi: 10.1080/15548627.2024.2396792. Epub 2024 Sep 20. Autophagy. 2025. PMID: 39245437 Free PMC article.
-
Structural basis for TNIP1 binding to FIP200 during mitophagy.J Biol Chem. 2024 Aug;300(8):107605. doi: 10.1016/j.jbc.2024.107605. Epub 2024 Jul 25. J Biol Chem. 2024. PMID: 39059492 Free PMC article.
-
A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1): a novel modulator of mitochondrial autophagy.Am J Physiol Cell Physiol. 2023 Feb 1;324(2):C339-C352. doi: 10.1152/ajpcell.00493.2022. Epub 2022 Nov 28. Am J Physiol Cell Physiol. 2023. PMID: 36440857 Free PMC article.
References
-
- Abudu, Y.P., Shrestha B.K., Zhang W., Palara A., Brenne H.B., Larsen K.B., Wolfson D.L., Dumitriu G., Øie C.I., Ahluwalia B.S., et al. . 2021. SAMM50 acts with p62 in piecemeal basal- and OXPHOS-induced mitophagy of SAM and MICOS components. J. Cell Biol. 220:220. 10.1083/jcb.202009092 - DOI - PMC - PubMed
-
- Akimov, V., Barrio-Hernandez I., Hansen S.V.F., Hallenborg P., Pedersen A.-K., Bekker-Jensen D.B., Puglia M., Christensen S.D.K., Vanselow J.T., Nielsen M.M., et al. . 2018. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25:631–640. 10.1038/s41594-018-0084-y - DOI - PubMed
-
- Alemu, E.A., Lamark T., Torgersen K.M., Birgisdottir A.B., Larsen K.B., Jain A., Olsvik H., Øvervatn A., Kirkin V., and Johansen T.. 2012. ATG8 family proteins act as scaffolds for assembly of the ULK complex: Sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 287:39275–39290. 10.1074/jbc.M112.378109 - DOI - PMC - PubMed
-
- Allanore, Y., Saad M., Dieudé P., Avouac J., Distler J.H., Amouyel P., Matucci-Cerinic M., Riemekasten G., Airo P., Melchers I., et al. . 2011. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 7:e1002091. 10.1371/journal.pgen.1002091 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
