

The landscape of therapeutic vulnerabilities in EGFR inhibitor osimertinib drug tolerant persister cells
- PMID: 36575215
- PMCID: PMC9794691
- DOI: 10.1038/s41698-022-00337-w
The landscape of therapeutic vulnerabilities in EGFR inhibitor osimertinib drug tolerant persister cells
Abstract
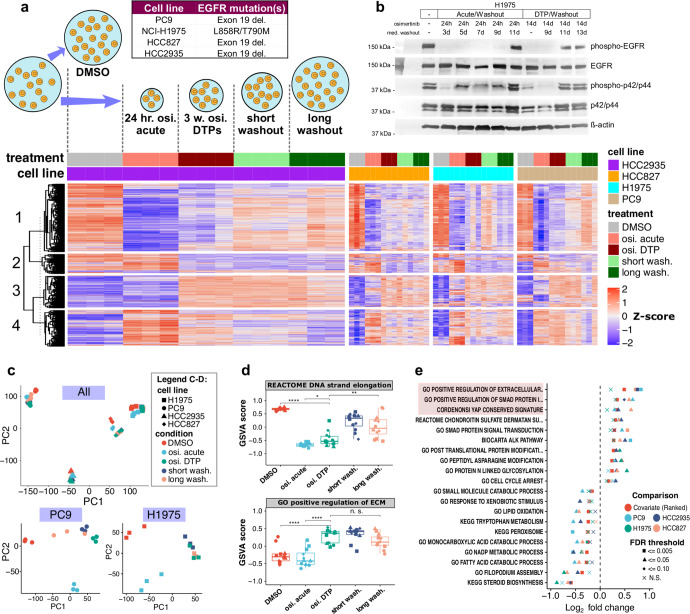
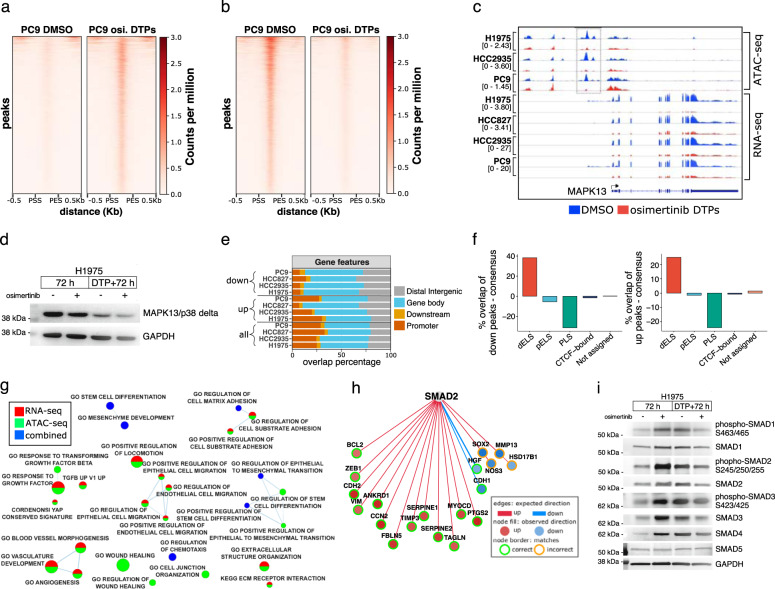
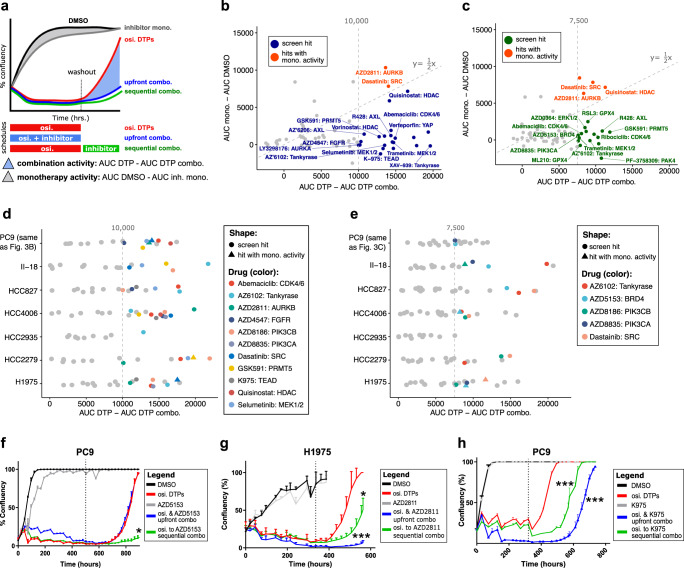
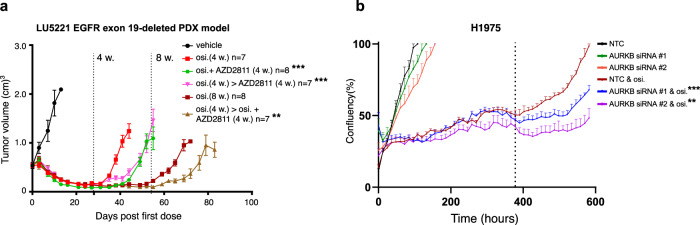
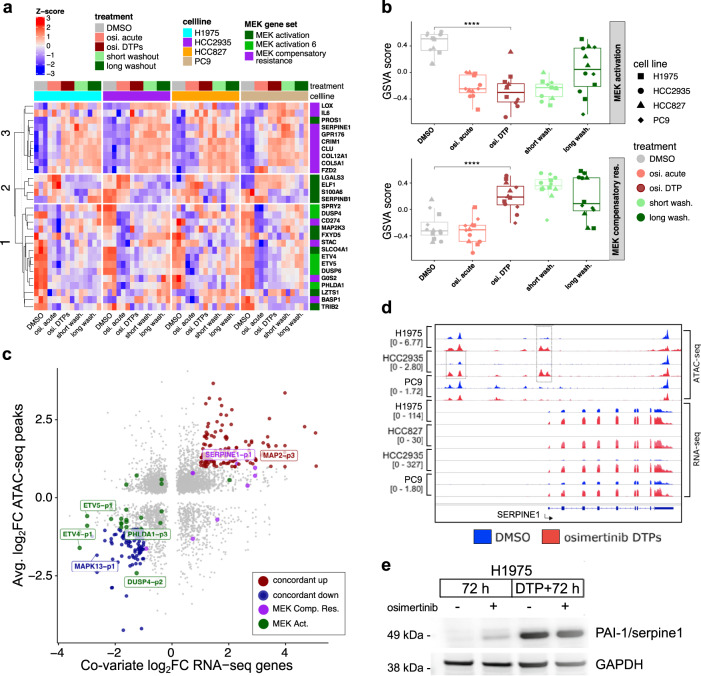
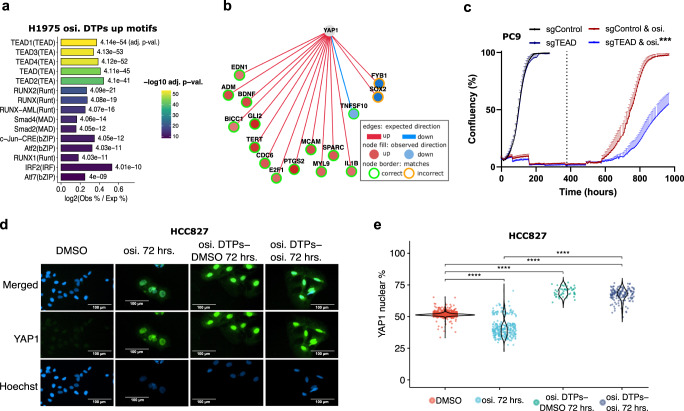
Third-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs), including osimertinib, an irreversible EGFR-TKI, are important treatments for non-small cell lung cancer with EGFR-TKI sensitizing or EGFR T790M resistance mutations. While patients treated with osimertinib show clinical benefit, disease progression and drug resistance are common. Emergence of de novo acquired resistance from a drug tolerant persister (DTP) cell population is one mechanism proposed to explain progression on osimertinib and other targeted cancer therapies. Here we profiled osimertinib DTPs using RNA-seq and ATAC-seq to characterize the features of these cells and performed drug screens to identify therapeutic vulnerabilities. We identified several vulnerabilities in osimertinib DTPs that were common across models, including sensitivity to MEK, AURKB, BRD4, and TEAD inhibition. We linked several of these vulnerabilities to gene regulatory changes, for example, TEAD vulnerability was consistent with evidence of Hippo pathway turning off in osimertinib DTPs. Last, we used genetic approaches using siRNA knockdown or CRISPR knockout to validate AURKB, BRD4, and TEAD as the direct targets responsible for the vulnerabilities observed in the drug screen.
© 2022. The Author(s).
Conflict of interest statement
All authors are or they were employees of AstraZeneca, and all authors held stock interests in AstraZeneca at the time this study was conducted.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
