

Lack of evidence for increased transcriptional noise in aged tissues
- PMID: 36576247
- PMCID: PMC9934862
- DOI: 10.7554/eLife.80380
Lack of evidence for increased transcriptional noise in aged tissues
Abstract
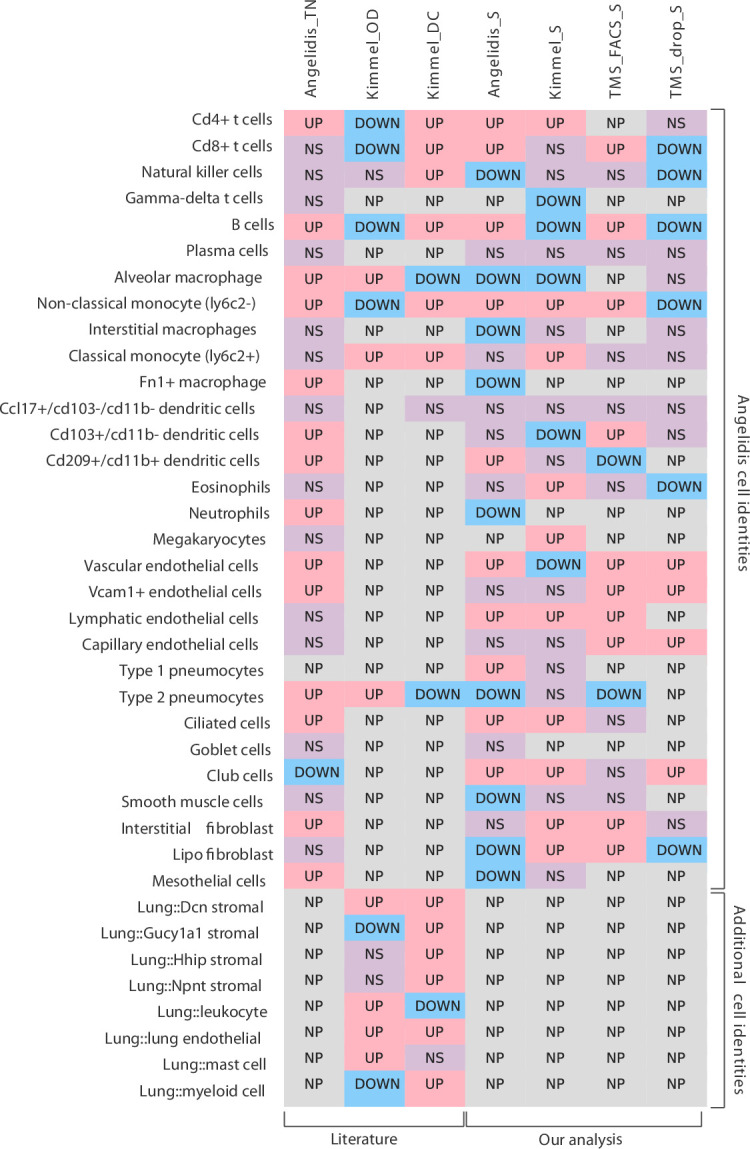
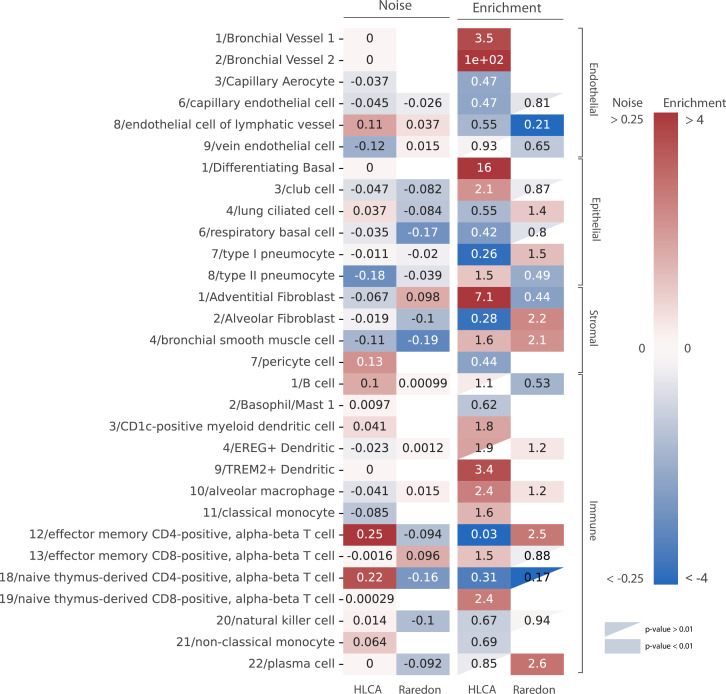
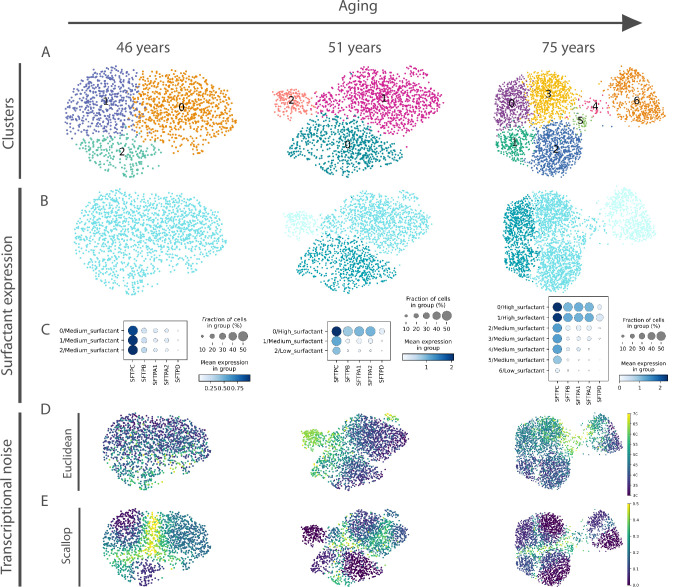
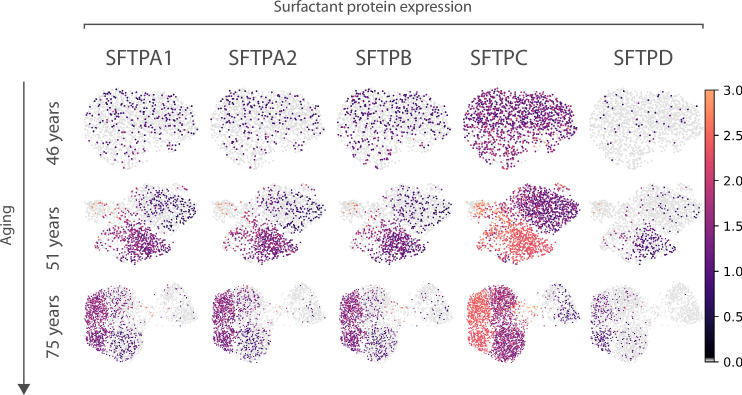
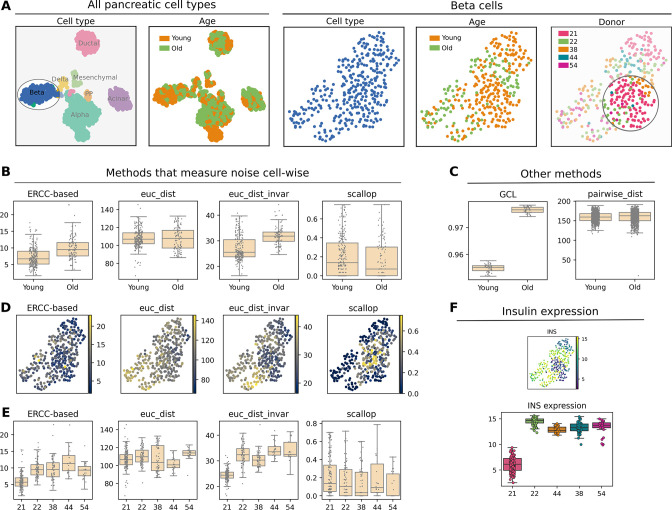
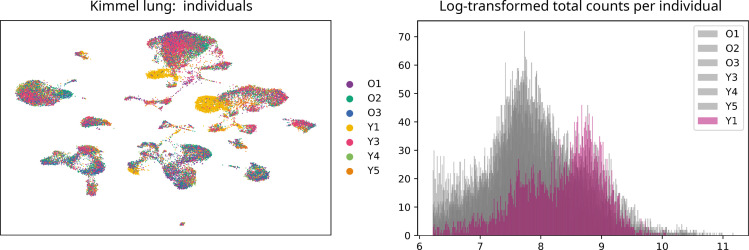
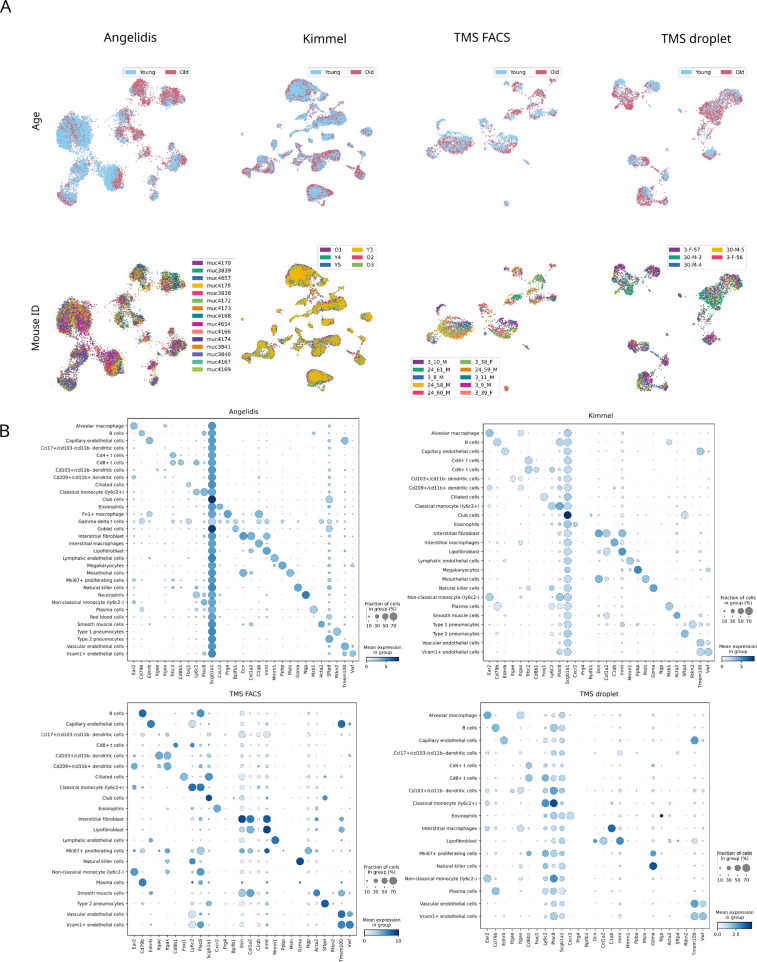
Aging is often associated with a loss of cell type identity that results in an increase in transcriptional noise in aged tissues. If this phenomenon reflects a fundamental property of aging remains an open question. Transcriptional changes at the cellular level are best detected by single-cell RNA sequencing (scRNAseq). However, the diverse computational methods used for the quantification of age-related loss of cellular identity have prevented reaching meaningful conclusions by direct comparison of existing scRNAseq datasets. To address these issues we created Decibel, a Python toolkit that implements side-to-side four commonly used methods for the quantification of age-related transcriptional noise in scRNAseq data. Additionally, we developed Scallop, a novel computational method for the quantification of membership of single cells to their assigned cell type cluster. Cells with a greater Scallop membership score are transcriptionally more stable. Application of these computational tools to seven aging datasets showed large variability between tissues and datasets, suggesting that increased transcriptional noise is not a universal hallmark of aging. To understand the source of apparent loss of cell type identity associated with aging, we analyzed cell type-specific changes in transcriptional noise and the changes in cell type composition of the mammalian lung. No robust pattern of cell type-specific transcriptional noise alteration was found across aging lung datasets. In contrast, age-associated changes in cell type composition of the lung were consistently found, particularly of immune cells. These results suggest that claims of increased transcriptional noise of aged tissues should be reformulated.
Keywords: aging; cell identity; computational biology; human; lung; mouse; single-cell RNA sequencing; systems biology; transcription; variability.
Plain language summary
The human body contains hundreds of different cell types which vary greatly in shape and size despite all sharing the same genetic material. This is because each cell switches on, or ‘expresses’, a unique set of genes that gives them a specific identity, such as becoming a nerve or a muscle cell. Recent studies have shown that cells in some tissues tend to lose their identity with age, and activate some of the genes that define them less strongly. This results in seemingly identical cells expressing the same genes in a more variable way, a phenomenon commonly referred to as noise. A technique called single-cell RNA sequencing is typically used to measure the activity of genes in individual cells, and has been used to study the role of noise in a wide range of aging tissues. However, the results of these studies have been analyzed using different computational methods, making it difficult to make comparisons between tissues and organisms. This has led to an ongoing debate about whether increased noise is a signature feature of aging, and if it is experienced throughout the body or restricted to certain cell types. To overcome this, Ibáñez-Solé, Ascensión et al. developed two new computational tools for analyzing noise and changes in cell identity: these were then applied to seven unique sequencing datasets which had been collected from various tissues in humans and mice at different ages. While there were some differences in the level of noise between young and old cells, these changes were not consistent across tissues and organisms. In contrast, other features associated with aging were consistently found in each of the sequencing datasets. The role of noise in aging has been gaining increasingly more attention in the scientific literature. However, the findings of Ibáñez-Solé, Ascensión et al. suggest that this phenomenon is not a hallmark of the aging process, and that the field should focus on other factors that reduce the health of tissues and cells as organisms get older. The computational approach they developed could also be used to evaluate the role of noise in other contexts, such as certain diseases.
© 2022, Ibañez-Solé, Ascensión et al.
Conflict of interest statement
OI, AA, MA, AI No competing interests declared
Figures
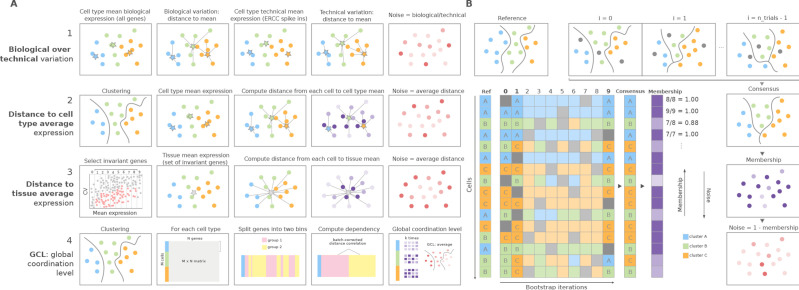
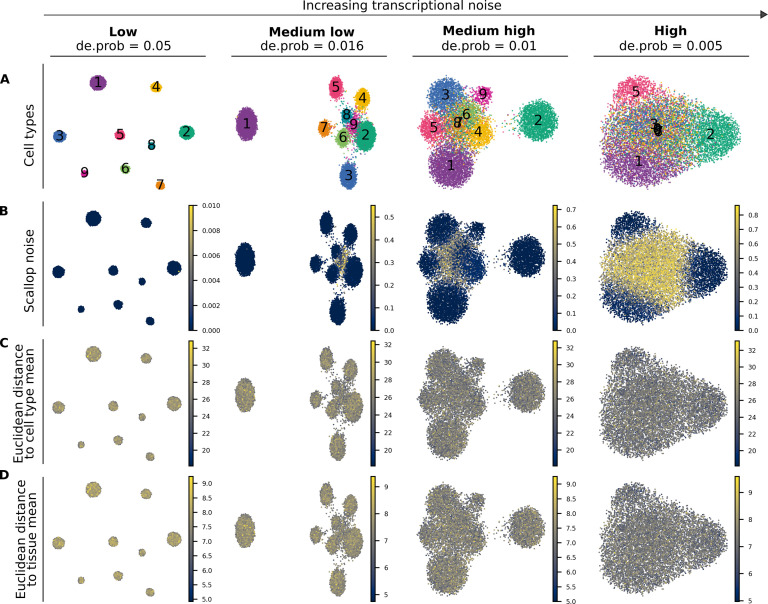
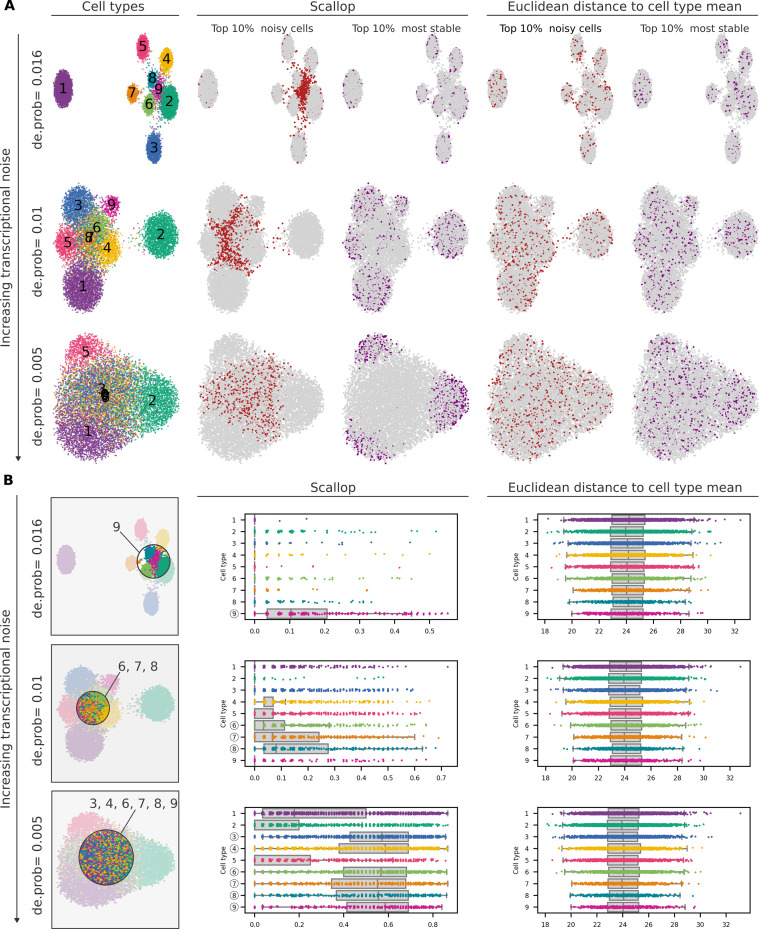
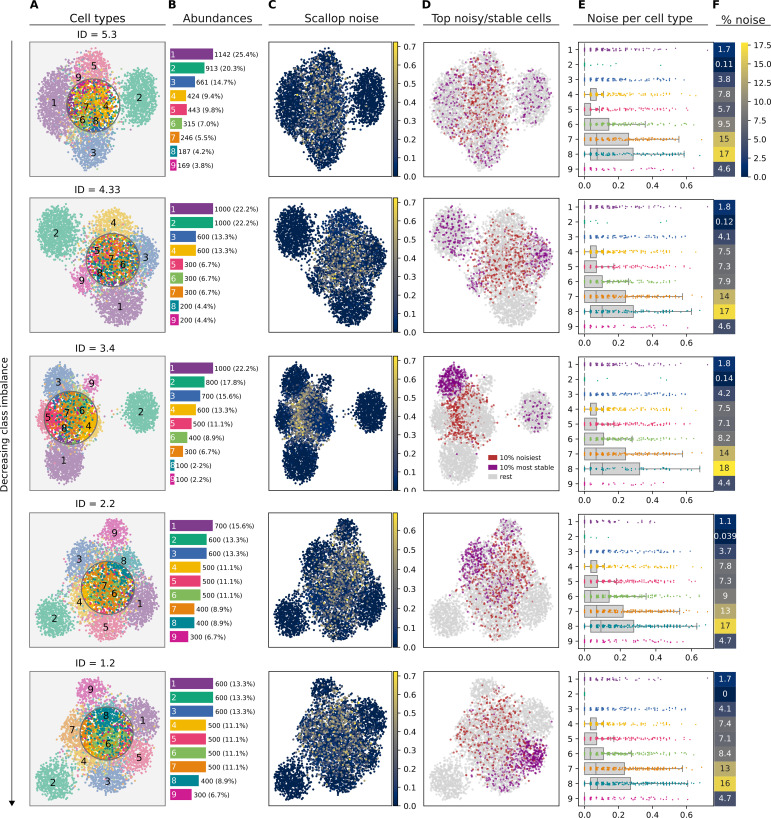
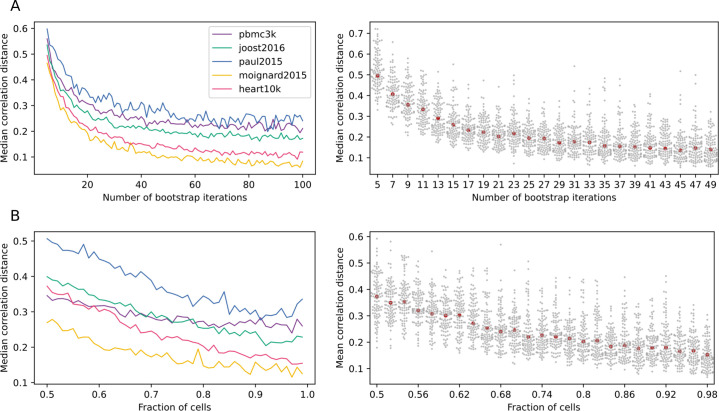
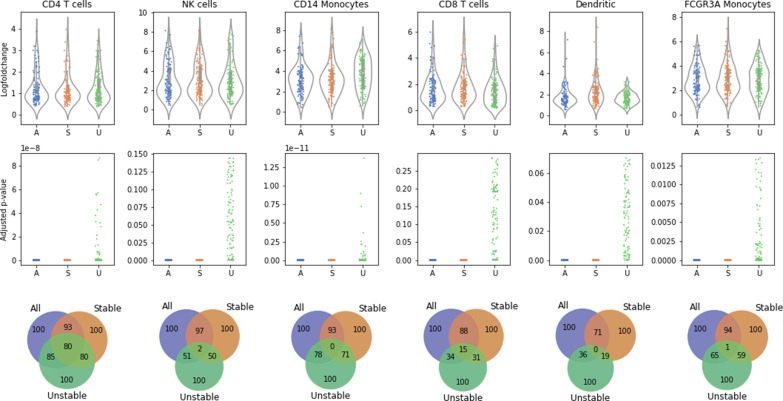
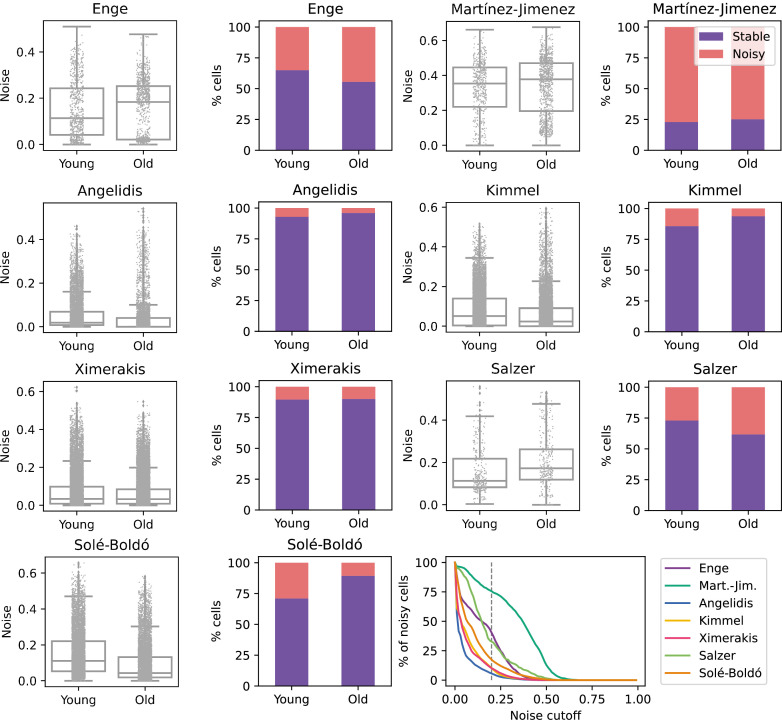
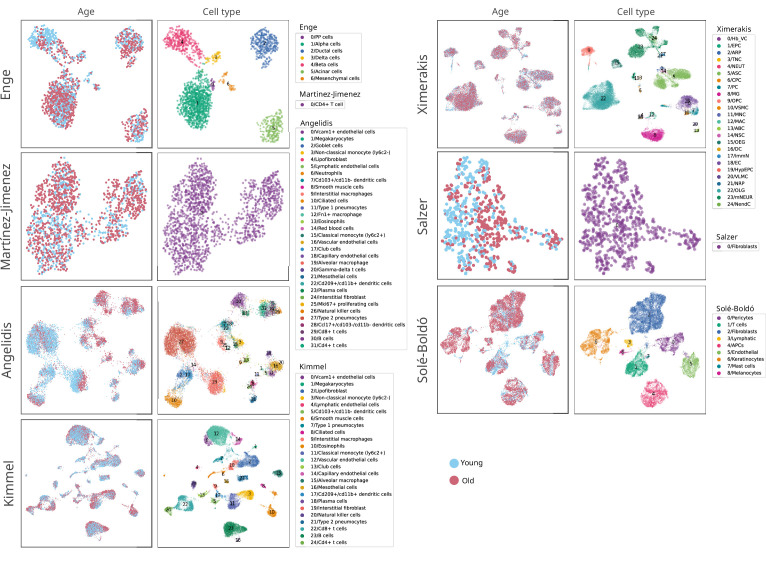
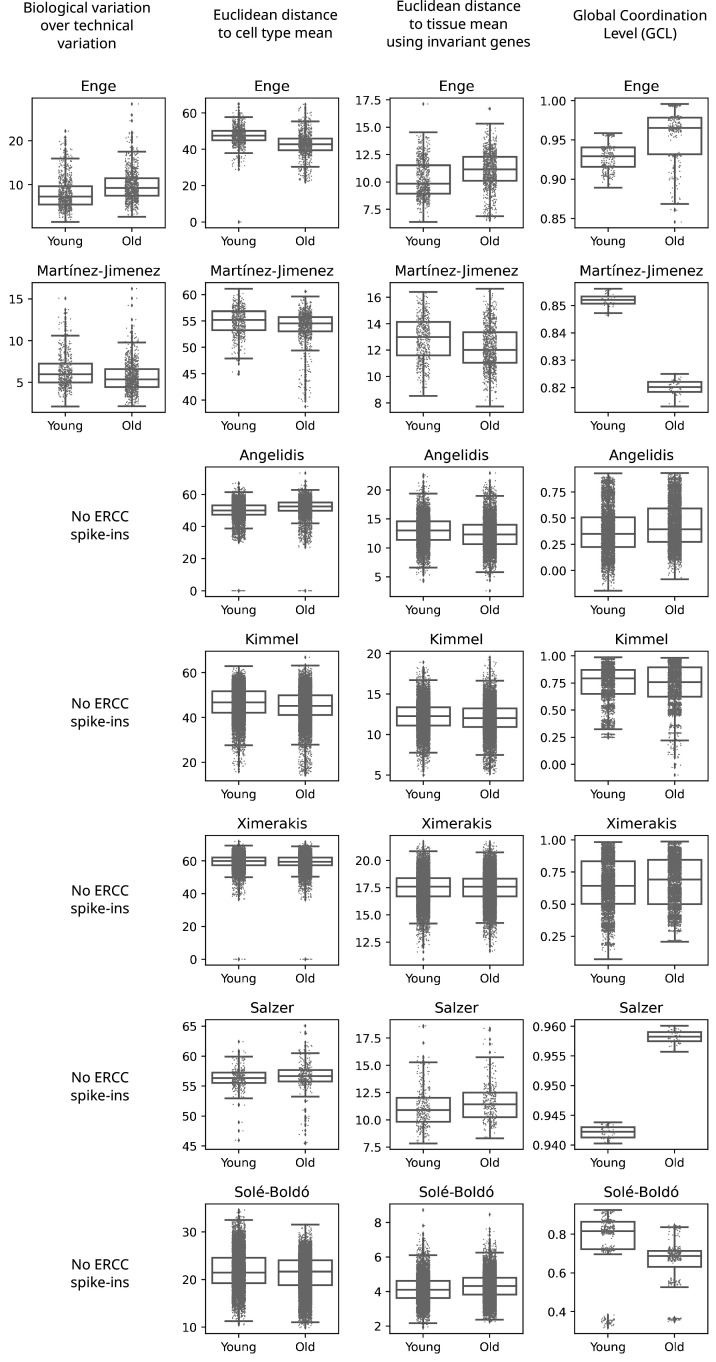
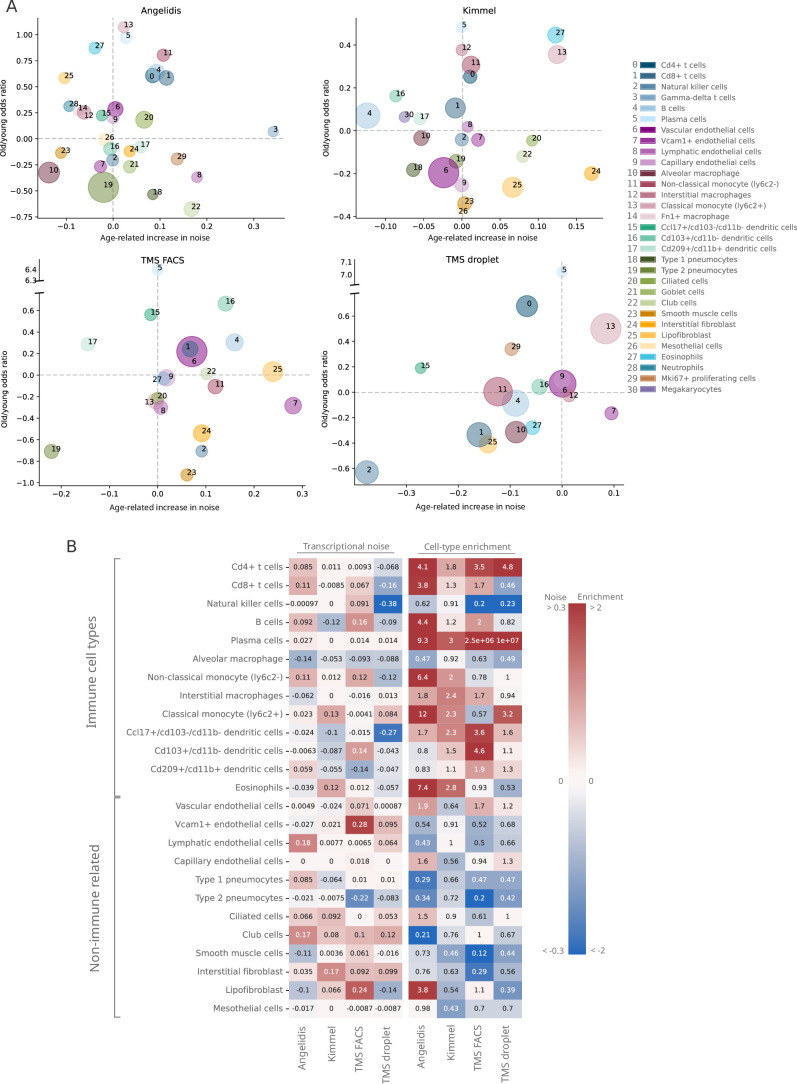
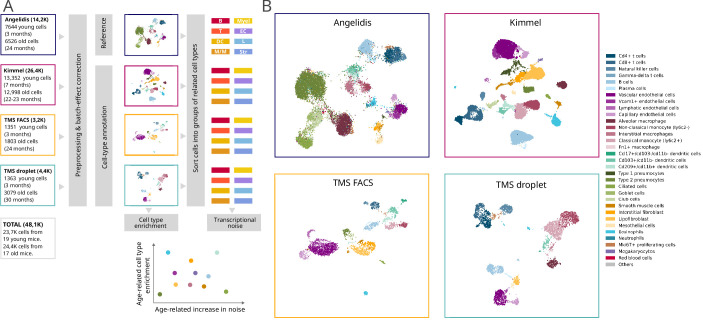
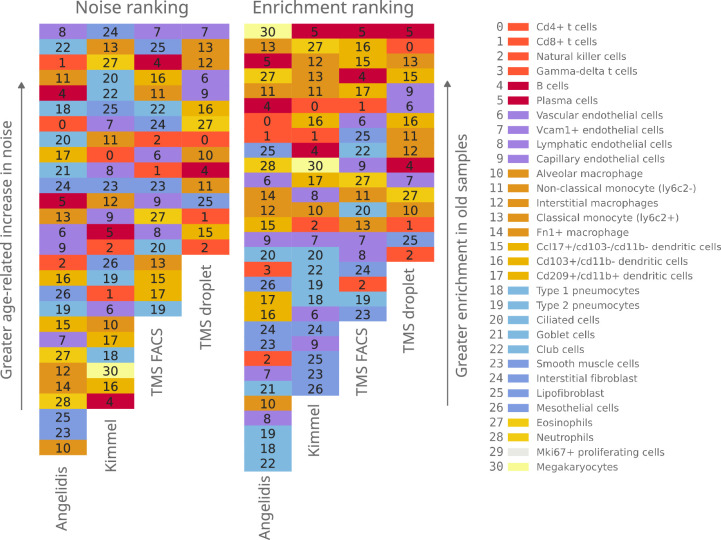

References
-
- Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom T-M, Nagendran M, Desai T, Eickelberg O, Mann M, Theis FJ, Schiller HB. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nature Communications. 2019;10:963. doi: 10.1038/s41467-019-08831-9. - DOI - PMC - PubMed
-
- Blondel VD, Guillaume JL, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics. 2008;1:10008. doi: 10.1088/1742-5468/2008/10/P10008. - DOI
-
- Cagan A, Baez-Ortega A, Brzozowska N, Abascal F, Coorens THH, Sanders MA, Lawson ARJ, Harvey LMR, Bhosle S, Jones D, Alcantara RE, Butler TM, Hooks Y, Roberts K, Anderson E, Lunn S, Flach E, Spiro S, Januszczak I, Wrigglesworth E, Jenkins H, Dallas T, Masters N, Perkins MW, Deaville R, Druce M, Bogeska R, Milsom MD, Neumann B, Gorman F, Constantino-Casas F, Peachey L, Bochynska D, Smith ESJ, Gerstung M, Campbell PJ, Murchison EP, Stratton MR, Martincorena I. Somatic mutation rates scale with lifespan across mammals. Nature. 2022;604:517–524. doi: 10.1038/s41586-022-04618-z. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- figshare/10.6084/m9.figshare.20402817.v1
LinkOut - more resources
Full Text Sources
Medical
