

A choline-releasing glycerophosphodiesterase essential for phosphatidylcholine biosynthesis and blood stage development in the malaria parasite
- PMID: 36576255
- PMCID: PMC9886279
- DOI: 10.7554/eLife.82207
A choline-releasing glycerophosphodiesterase essential for phosphatidylcholine biosynthesis and blood stage development in the malaria parasite
Abstract
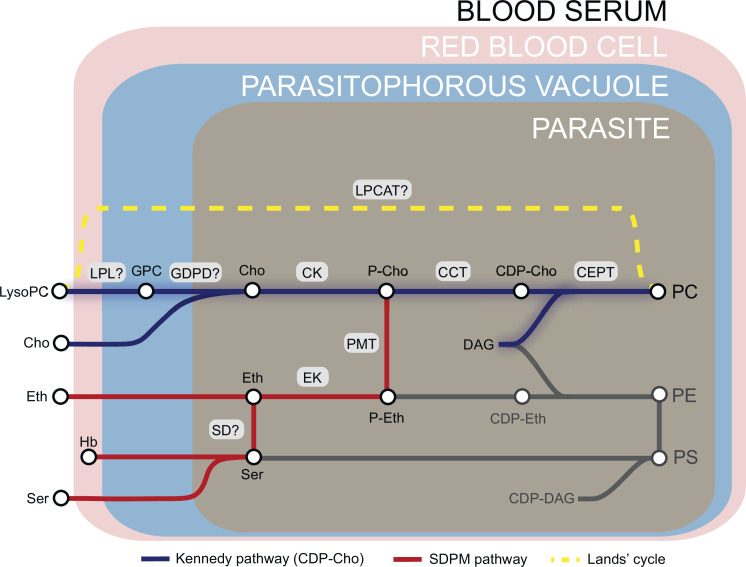
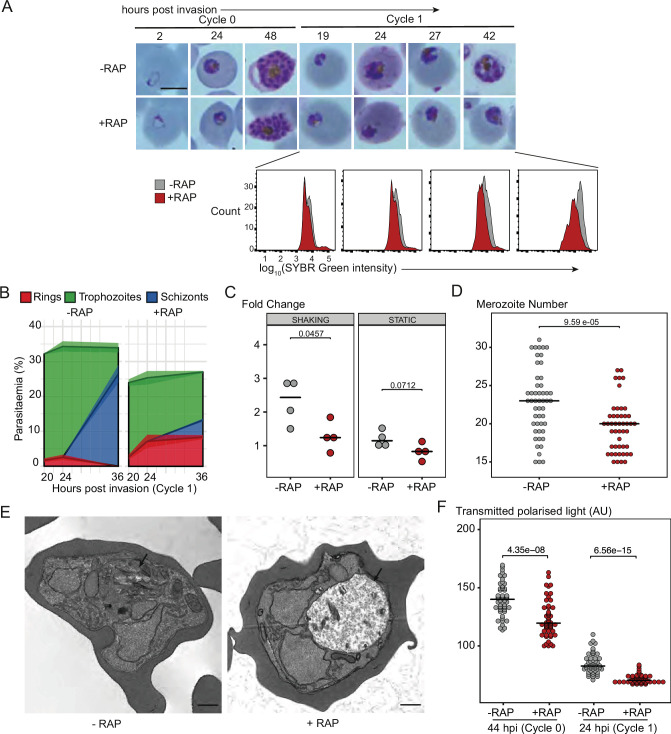
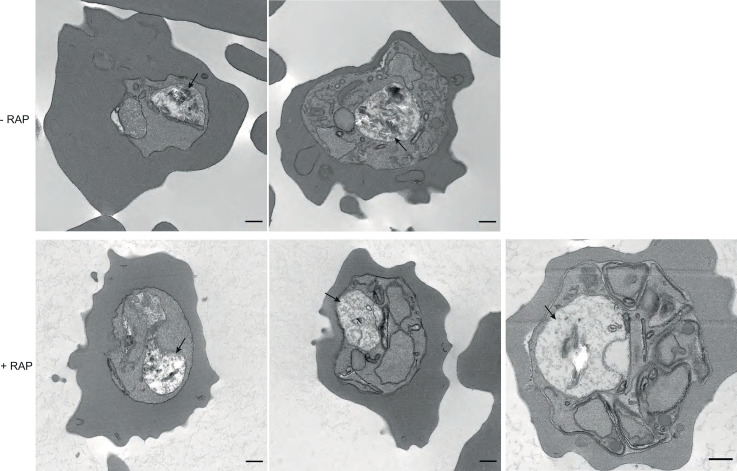
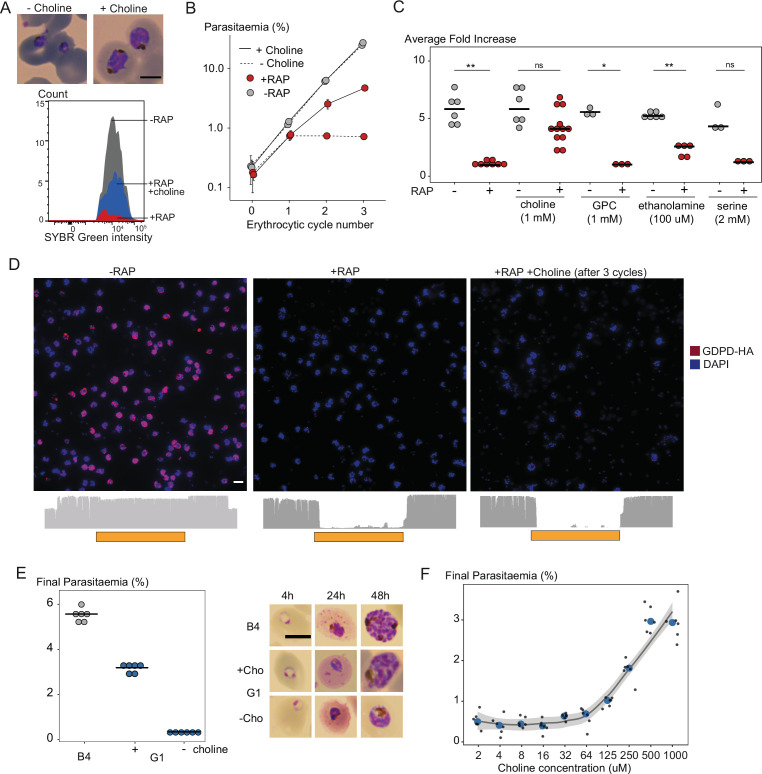
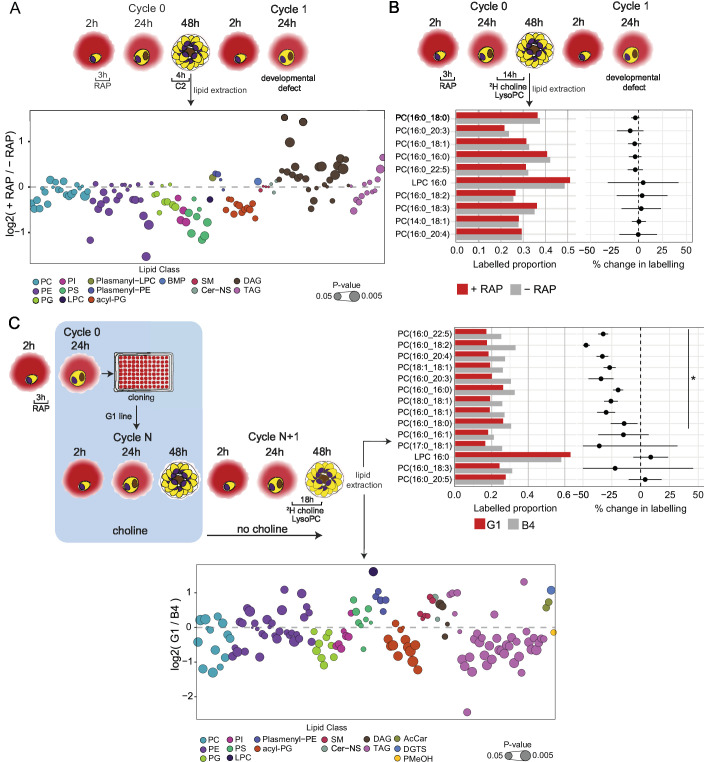
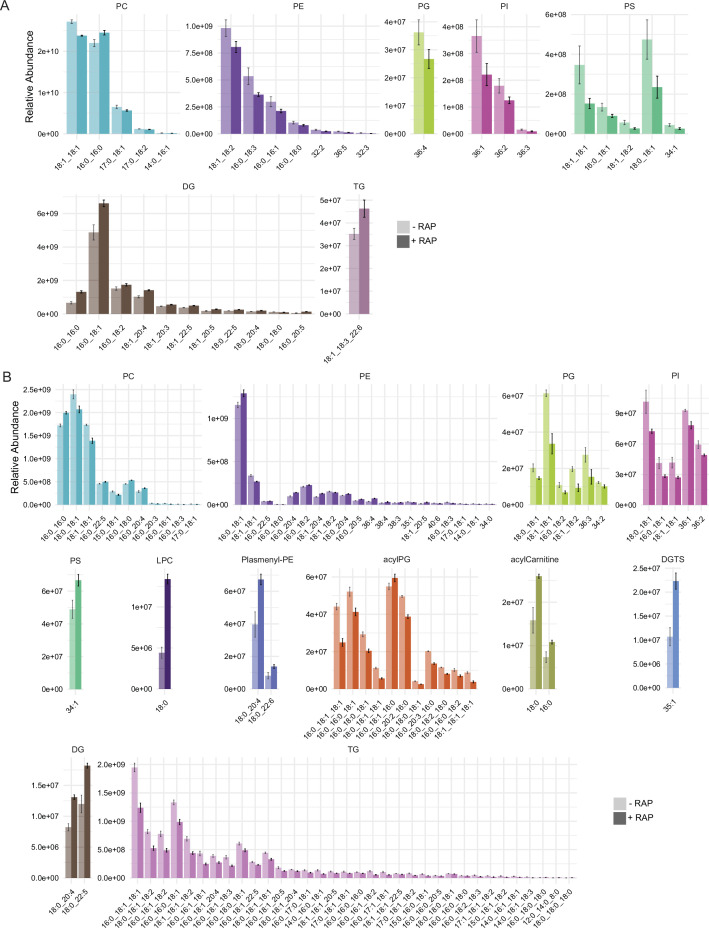
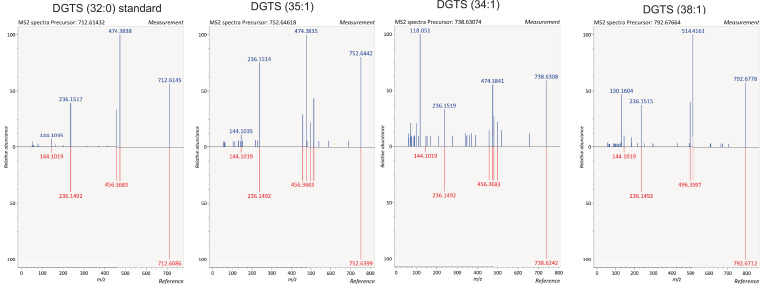
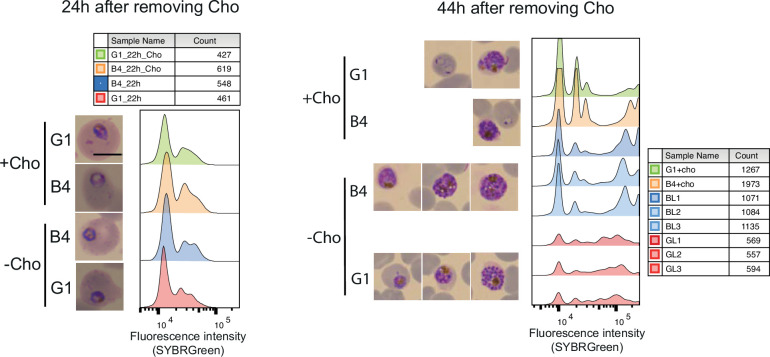
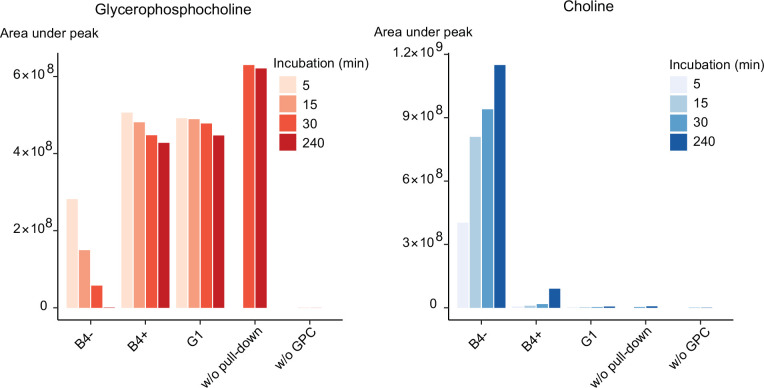
The malaria parasite Plasmodium falciparum synthesizes significant amounts of phospholipids to meet the demands of replication within red blood cells. De novo phosphatidylcholine (PC) biosynthesis via the Kennedy pathway is essential, requiring choline that is primarily sourced from host serum lysophosphatidylcholine (lysoPC). LysoPC also acts as an environmental sensor to regulate parasite sexual differentiation. Despite these critical roles for host lysoPC, the enzyme(s) involved in its breakdown to free choline for PC synthesis are unknown. Here, we show that a parasite glycerophosphodiesterase (PfGDPD) is indispensable for blood stage parasite proliferation. Exogenous choline rescues growth of PfGDPD-null parasites, directly linking PfGDPD function to choline incorporation. Genetic ablation of PfGDPD reduces choline uptake from lysoPC, resulting in depletion of several PC species in the parasite, whilst purified PfGDPD releases choline from glycerophosphocholine in vitro. Our results identify PfGDPD as a choline-releasing glycerophosphodiesterase that mediates a critical step in PC biosynthesis and parasite survival.
Keywords: P. falciparum; Plasmodium; biochemistry; chemical biology; glycerophosphodiesterase; infectious disease; lysophosphatidylcholine; malaria; microbiology; phosphatidylcholine synthesis.
Plain language summary
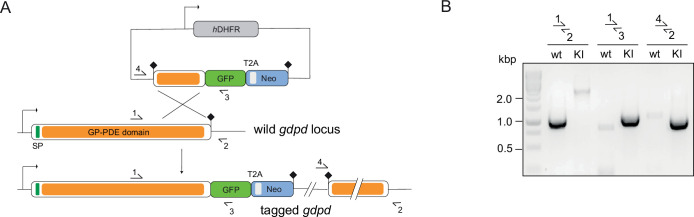
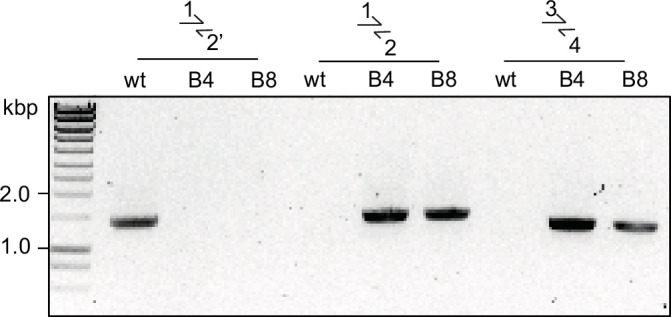
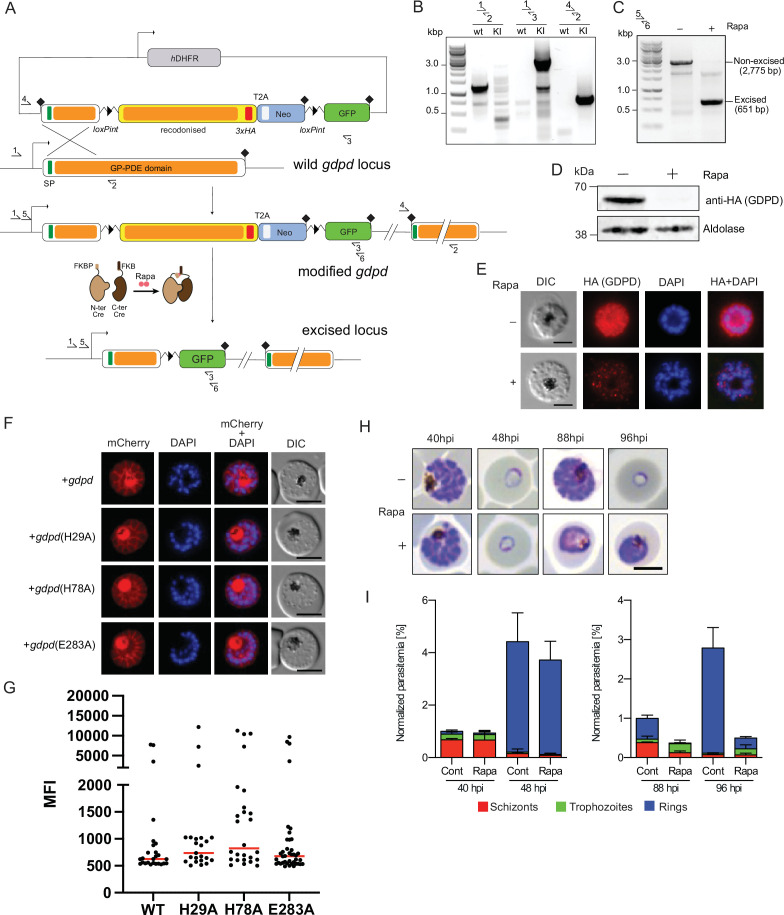
Malaria kills over half a million people every year worldwide. A single-celled parasite called Plasmodium falciparum is responsible for the most lethal form of the disease. This malaria-causing agent is carried by mosquitos which transmit the parasite to humans through their bite. Once in the bloodstream, the parasite enters red blood cells and starts to replicate so it can go on to infect other cells. Like our cells, P. falciparum is surrounded by a membrane, and further membranes surround a number of its internal compartments. To make these protective coats, the parasite has to gather a nutrient called choline to form an important building block in the membrane. The parasite gets most of its choline by absorbing and digesting a molecule known as lysoPC found in the bloodstream of its host. However, it was unclear precisely how the parasite achieves this. To address this question, Ramaprasad, Burda et al. used genetic and metabolomic approaches to study how P. falciparum breaks down lysoPC. The experiments found that mutant parasites that are unable to make an enzyme called GDPD were able to infect red blood cells, but failed to grow properly once inside the cells. The mutant parasites took up less choline and, as a result, also made fewer membrane building blocks. The team were able to rescue the mutant parasites by supplying them with large quantities of choline, which allowed them to resume growing. Taken together, the findings of Ramaprasad, Burda et al. suggest that P. falciparum uses GDPD to extract choline from lysoPC when it is living in red blood cells. More and more P. falciparum parasites are becoming resistant to many of the drugs currently being used to treat malaria. One solution is to develop new therapies that target different molecules in the parasite. Since it performs such a vital role, GDPD may have the potential to be a future drug target.
© 2022, Ramaprasad, Burda et al.
Conflict of interest statement
AR, PB, EC, AS, SP, CW, FH, JM, LC, TG, MB No competing interests declared
Figures

Similar articles
-
Genetic and chemical validation of Plasmodium falciparum aminopeptidase PfA-M17 as a drug target in the hemoglobin digestion pathway.Elife. 2022 Sep 13;11:e80813. doi: 10.7554/eLife.80813. Elife. 2022. PMID: 36097817 Free PMC article.
-
The malaria parasite sheddase SUB2 governs host red blood cell membrane sealing at invasion.Elife. 2020 Dec 8;9:e61121. doi: 10.7554/eLife.61121. Elife. 2020. PMID: 33287958 Free PMC article.
-
The patatin-like phospholipase PfPNPLA2 is involved in the mitochondrial degradation of phosphatidylglycerol during Plasmodium falciparum blood stage development.Front Cell Infect Microbiol. 2023 Nov 20;13:997245. doi: 10.3389/fcimb.2023.997245. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 38089812 Free PMC article.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
Of membranes and malaria: phospholipid asymmetry in Plasmodium falciparum-infected red blood cells.Cell Mol Life Sci. 2021 May;78(10):4545-4561. doi: 10.1007/s00018-021-03799-6. Epub 2021 Mar 13. Cell Mol Life Sci. 2021. PMID: 33713154 Free PMC article. Review.
Cited by
-
A scaleable inducible knockout system for studying essential gene function in the malaria parasite.Nucleic Acids Res. 2025 Feb 8;53(4):gkae1274. doi: 10.1093/nar/gkae1274. Nucleic Acids Res. 2025. PMID: 39739757 Free PMC article.
-
P. falciparum Invasion and Erythrocyte Aging.Cells. 2024 Feb 12;13(4):334. doi: 10.3390/cells13040334. Cells. 2024. PMID: 38391947 Free PMC article. Review.
-
Photoaffinity probe-based antimalarial target identification of artemisinin in the intraerythrocytic developmental cycle of Plasmodium falciparum.Imeta. 2024 Feb 19;3(2):e176. doi: 10.1002/imt2.176. eCollection 2024 Apr. Imeta. 2024. PMID: 38882489 Free PMC article.
-
Three-dimensional reconstruction of rat sperm using volume electron microscopy.Acta Biochim Biophys Sin (Shanghai). 2024 Sep 3;56(11):1699-1705. doi: 10.3724/abbs.2024144. Acta Biochim Biophys Sin (Shanghai). 2024. PMID: 39243140 Free PMC article.
-
Host metabolomic responses in recurrent P. vivax malaria.Sci Rep. 2024 Mar 27;14(1):7249. doi: 10.1038/s41598-024-54231-5. Sci Rep. 2024. PMID: 38538661 Free PMC article.
References
-
- Abagyan R, Totrov M, Kuznetsov D. ICM?a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. Journal of Computational Chemistry. 1994;15:488–506. doi: 10.1002/jcc.540150503. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
