

Basis Set Limit CCSD(T) Energies for Extended Molecules via a Reduced-Cost Explicitly Correlated Approach
- PMID: 36576419
- PMCID: PMC9835832
- DOI: 10.1021/acs.jctc.2c01031
Basis Set Limit CCSD(T) Energies for Extended Molecules via a Reduced-Cost Explicitly Correlated Approach
Abstract
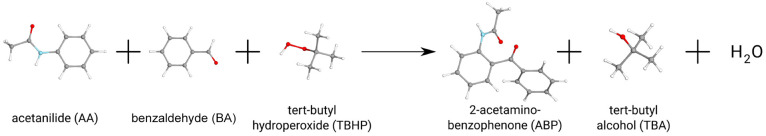
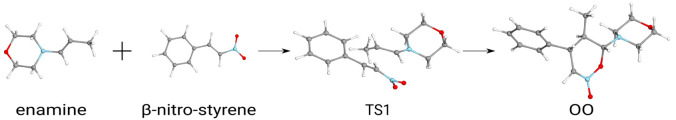
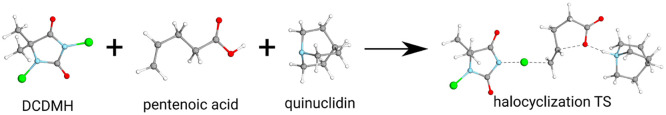
Several approximations are introduced and tested to reduce the computational expenses of the explicitly correlated coupled-cluster singles and doubles with perturbative triples [CCSD(T)] method for both closed and open-shell species. First, the well-established frozen natural orbital (FNO) technique is adapted to explicitly correlated CC approaches. Second, our natural auxiliary function (NAF) scheme is employed to reduce the size of the auxiliary basis required for the density fitting approximation regularly used in explicitly correlated calculations. Third, a new approach, termed the natural auxiliary basis (NAB) approximation, is proposed to decrease the size of the auxiliary basis needed for the expansion of the explicitly correlated geminals. The performance of the above approximations and that of the combined FNO-NAF-NAB approach are tested for atomization and reaction energies. Our results show that overall speedups of 7-, 5-, and 3-times can be achieved with double-, triple-, and quadruple-ζ basis sets, respectively, without any loss in accuracy. The new method can provide, e.g., reaction energies and barrier heights well within chemical accuracy for molecules with more than 40 atoms within a few days using a few dozen processor cores, and calculations with 50+ atoms are still feasible. These routinely affordable computations considerably extend the reach of explicitly correlated CCSD(T).
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Basis Set Limit of CCSD(T) Energies: Explicit Correlation Versus Density-Based Basis-Set Correction.J Chem Theory Comput. 2023 Nov 28;19(22):8210-8222. doi: 10.1021/acs.jctc.3c00979. Epub 2023 Nov 11. J Chem Theory Comput. 2023. PMID: 37950703 Free PMC article.
-
Accurate Reduced-Cost CCSD(T) Energies: Parallel Implementation, Benchmarks, and Large-Scale Applications.J Chem Theory Comput. 2021 Feb 9;17(2):860-878. doi: 10.1021/acs.jctc.0c01077. Epub 2021 Jan 5. J Chem Theory Comput. 2021. PMID: 33400527 Free PMC article.
-
Simple coupled-cluster singles and doubles method with perturbative inclusion of triples and explicitly correlated geminals: The CCSD(T)R12 model.J Chem Phys. 2008 Jun 28;128(24):244113. doi: 10.1063/1.2939577. J Chem Phys. 2008. PMID: 18601323
-
Enabling Accurate and Large-Scale Explicitly Correlated CCSD(T) Computations via a Reduced-Cost and Parallel Implementation.J Chem Theory Comput. 2025 Mar 11;21(5):2432-2447. doi: 10.1021/acs.jctc.4c01777. Epub 2025 Feb 26. J Chem Theory Comput. 2025. PMID: 40008851 Free PMC article.
-
On the accuracy of explicitly correlated coupled-cluster interaction energies--have orbital results been beaten yet?J Chem Phys. 2012 Jul 21;137(3):034103. doi: 10.1063/1.4734597. J Chem Phys. 2012. PMID: 22830679
Cited by
-
Analytic Gradients for Density Fitting MP2 Using Natural Auxiliary Functions.J Phys Chem A. 2024 Aug 8;128(31):6566-6580. doi: 10.1021/acs.jpca.4c02822. Epub 2024 Jul 29. J Phys Chem A. 2024. PMID: 39074307 Free PMC article.
-
Accurate Structures and Spectroscopic Parameters of Guanine Tautomers in the Gas Phase by the Pisa Conventional and Explicitly Correlated Composite Schemes (PCS and PCS-F12).J Phys Chem A. 2023 Aug 17;127(32):6771-6778. doi: 10.1021/acs.jpca.3c03999. Epub 2023 Aug 3. J Phys Chem A. 2023. PMID: 37535450 Free PMC article.
-
Advancing Non-Atom-Centered Basis Methods for More Accurate Interaction Energies: Benchmarks and Large-Scale Applications.J Phys Chem A. 2024 Nov 28;128(47):10282-10298. doi: 10.1021/acs.jpca.4c04689. Epub 2024 Nov 18. J Phys Chem A. 2024. PMID: 39556045 Free PMC article.
-
Linear-Scaling Local Natural Orbital CCSD(T) Approach for Open-Shell Systems: Algorithms, Benchmarks, and Large-Scale Applications.J Chem Theory Comput. 2023 Nov 28;19(22):8166-8188. doi: 10.1021/acs.jctc.3c00881. Epub 2023 Nov 3. J Chem Theory Comput. 2023. PMID: 37921429 Free PMC article.
-
Basis Set Limit of CCSD(T) Energies: Explicit Correlation Versus Density-Based Basis-Set Correction.J Chem Theory Comput. 2023 Nov 28;19(22):8210-8222. doi: 10.1021/acs.jctc.3c00979. Epub 2023 Nov 11. J Chem Theory Comput. 2023. PMID: 37950703 Free PMC article.
References
-
- Purvis G. D. III; Bartlett R. J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910.10.1063/1.443164. - DOI
-
- Raghavachari K.; Trucks G. W.; Pople J. A.; Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479.10.1016/S0009-2614(89)87395-6. - DOI
-
- Adamowicz L.; Bartlett R. J. Optimized virtual orbital space for high-level correlated calculations. J. Chem. Phys. 1987, 86, 6314.10.1063/1.452468. - DOI
-
- Adamowicz L.; Bartlett R. J.; Sadlej J. Optimized virtual orbital space for high-level correlated calculations. II. Electric properties. J. Chem. Phys. 1988, 88, 5749.10.1063/1.454721. - DOI
-
- Sosa C.; Geertsen J.; Trucks G. W.; Bartlett R. J.; Franz J. A. Selection of the reduced virtual space for correlated calculations. An application to the energy and dipole moment of H2O. Chem. Phys. Lett. 1989, 159, 148.10.1016/0009-2614(89)87399-3. - DOI
LinkOut - more resources
Full Text Sources
