

On the lookout for influenza viruses in Italy during the 2021-2022 season: Along came A(H3N2) viruses with a new phylogenetic makeup of their hemagglutinin
- PMID: 36581046
- PMCID: PMC10194219
- DOI: 10.1016/j.virusres.2022.199033
On the lookout for influenza viruses in Italy during the 2021-2022 season: Along came A(H3N2) viruses with a new phylogenetic makeup of their hemagglutinin
Abstract
Aims: To assess influenza viruses (IVs) circulation and to evaluate A(H3N2) molecular evolution during the 2021-2022 season in Italy.
Materials and methods: 12,393 respiratory specimens (nasopharyngeal swabs or broncho-alveolar lavages) collected from in/outpatients with influenza illness in the period spanning from January 1, 2022 (week 2022-01) to May 31, 2022 (week 2022-22) were analysed to identify IV genome and were molecularly characterized by 12 laboratories throughout Italy. A(H3N2) evolution was studied by conducting an in-depth phylogenetic analysis of the hemagglutinin (HA) gene sequences. The predicted vaccine efficacy (pVE) of vaccine strain against circulating A(H3N2) viruses was estimated using the sequence-based Pepitope model.
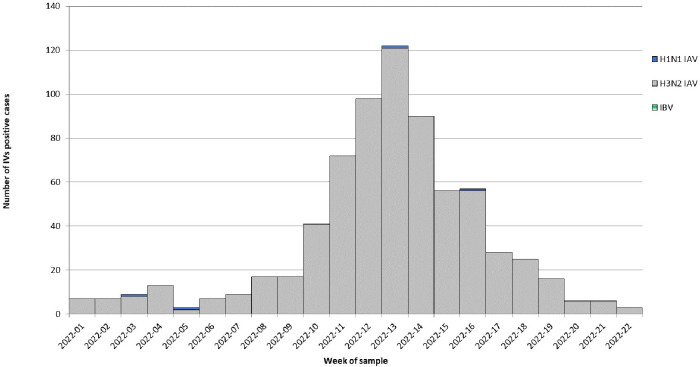
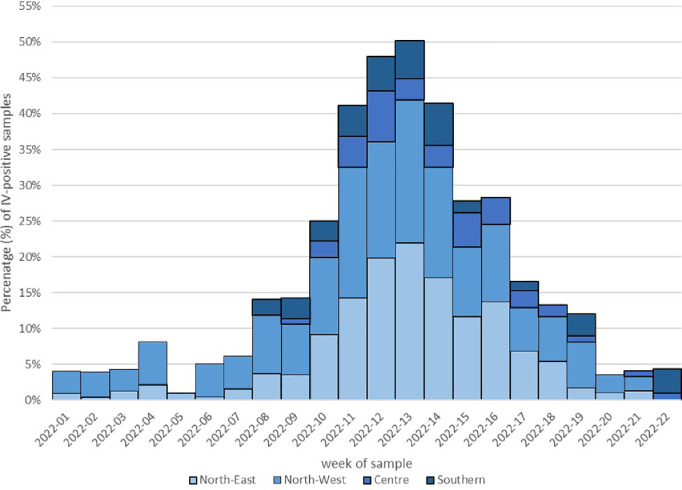
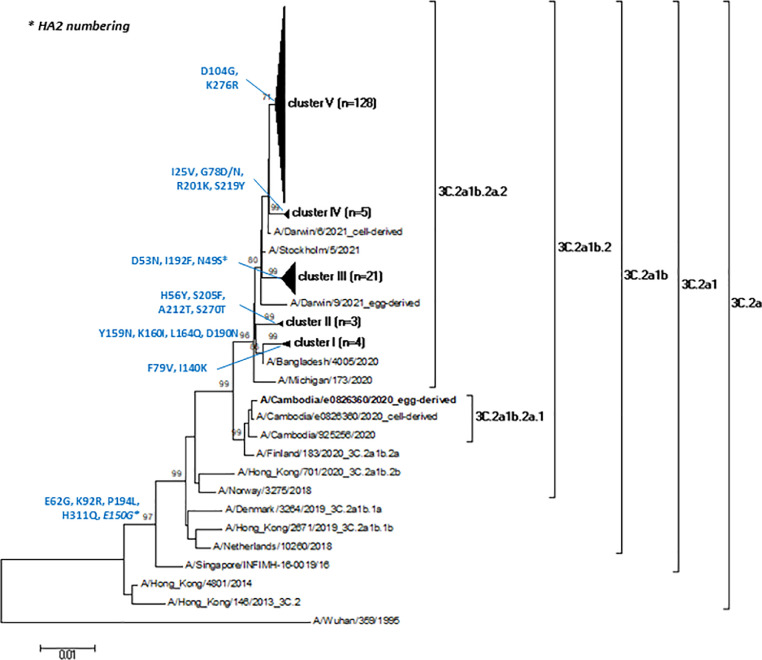
Results: The overall IV-positive rate was 7.2% (894/12,393), all were type A IVs. Almost all influenza A viruses (846/894; 94.6%) were H3N2 that circulated in Italy with a clear epidemic trend, with 10% positivity rate threshold crossed for six consecutive weeks from week 2022-11 to week 2022-16. According to the phylogenetic analysis of a subset of A(H3N2) strains (n=161), the study HA sequences were distributed into five different genetic clusters, all of them belonging to the clade 3C.2a, sub-clade 3C.2a1 and the genetic subgroup 3C.2a1b.2a.2. The selective pressure analysis of A(H3N2) sequences showed evidence of diversifying selection particularly in the amino acid position 156. The comparison between the predicted amino acid sequence of the 2021-2022 vaccine strain (A/Cambodia/e0826360/2020) and the study strains revealed 65 mutations in 59 HA amino acid positions, including the substitution H156S and Y159N in antigenic site B, within major antigenic sites adjacent to the receptor-binding site, suggesting the presence of drifted strains. According to the sequence-based Pepitope model, antigenic site B was the dominant antigenic site and the p(VE) against circulating A(H3N2) viruses was estimated to be -28.9%.
Discussion and conclusion: After a long period of very low IV activity since public health control measures have been introduced to face COVID-19 pandemic, along came A(H3N2) with a new phylogenetic makeup. Although the delayed 2021-2022 influenza season in Italy was characterized by a significant reduction of the width of the epidemic curve and in the intensity of the influenza activity compared to historical data, a marked genetic diversity of the HA of circulating A(H3N2) strains was observed. The identification of the H156S and Y159N substitutions within the main antigenic sites of most HA sequences also suggested the circulation of drifted variants with respect to the 2021-2022 vaccine strain. Molecular surveillance plays a critical role in the influenza surveillance architecture and it has to be strengthened also at local level to timely assess vaccine effectiveness and detect novel strains with potential impact on public health.
Keywords: A(H3N2); Influenza; Phylogenetic analysis; Predicted vaccine analyses; Selective pressure analysis.
Copyright © 2022. Published by Elsevier B.V.
Conflict of interest statement
Conflict of interest Nothing to declare.
Figures
References
-
- Petrova VN, Russell CA. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018 Jan;16(1):47–60. - PubMed
-
- Word Health Organization (OMS). Influenza seasonal. https://www.who.int/health-topics/influenza-seasonal#tab=tab_1. Last Accessed: 29/11/2022.
-
- Weis W, Brown JH, Cusack S, et al. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature. 1988 Jun;333(6172):426–431. - PubMed
