

Impaired Human Cardiac Cell Development due to NOTCH1 Deficiency
- PMID: 36583388
- PMCID: PMC9852089
- DOI: 10.1161/CIRCRESAHA.122.321398
Impaired Human Cardiac Cell Development due to NOTCH1 Deficiency
Abstract
Background: NOTCH1 pathogenic variants are implicated in multiple types of congenital heart defects including hypoplastic left heart syndrome, where the left ventricle is underdeveloped. It is unknown how NOTCH1 regulates human cardiac cell lineage determination and cardiomyocyte proliferation. In addition, mechanisms by which NOTCH1 pathogenic variants lead to ventricular hypoplasia in hypoplastic left heart syndrome remain elusive.
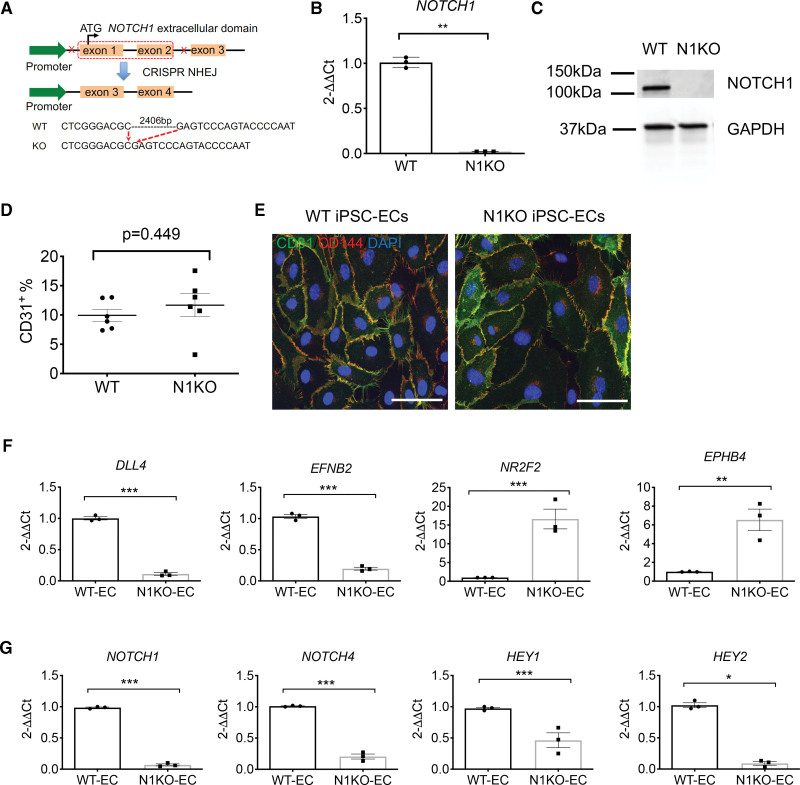
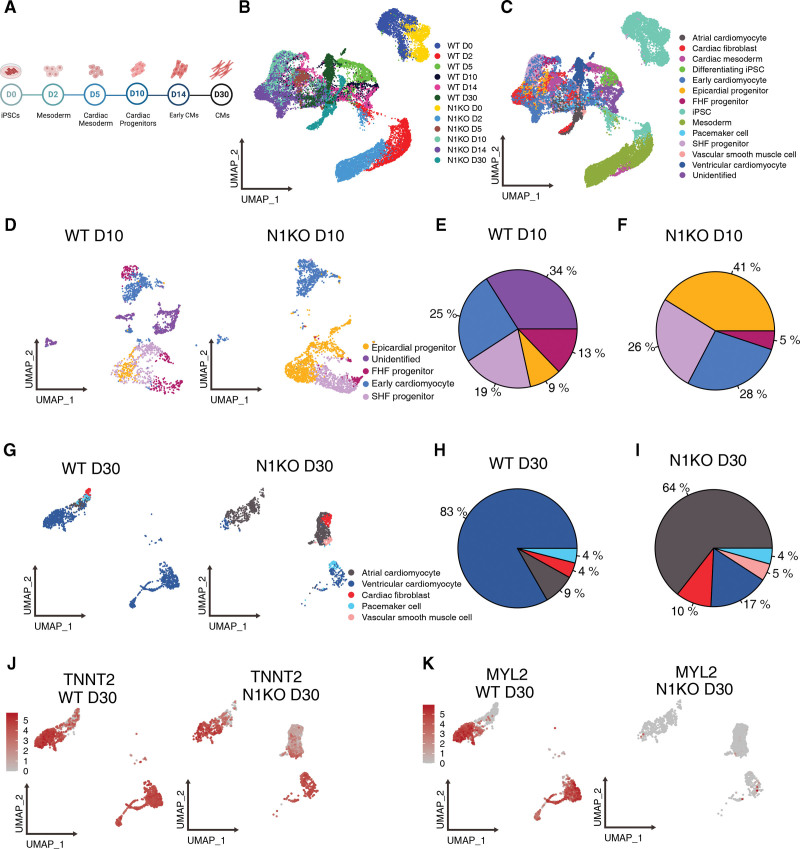
Methods: CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9 genome editing was utilized to delete NOTCH1 in human induced pluripotent stem cells. Cardiac differentiation was carried out by sequential modulation of WNT signaling, and NOTCH1 knockout and wild-type differentiating cells were collected at day 0, 2, 5, 10, 14, and 30 for single-cell RNA-seq.
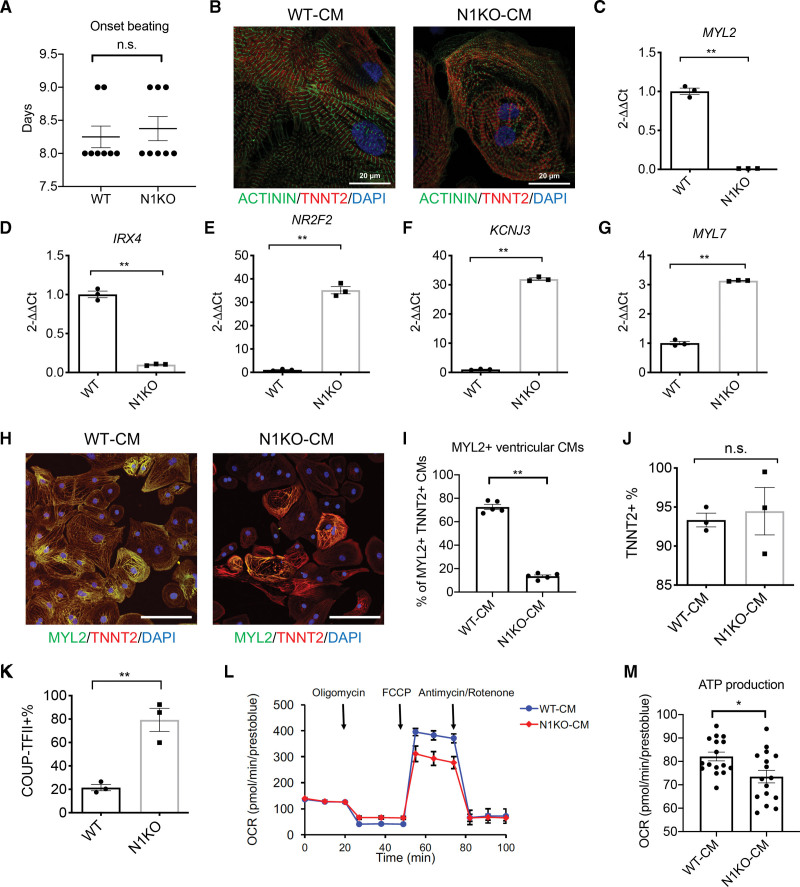
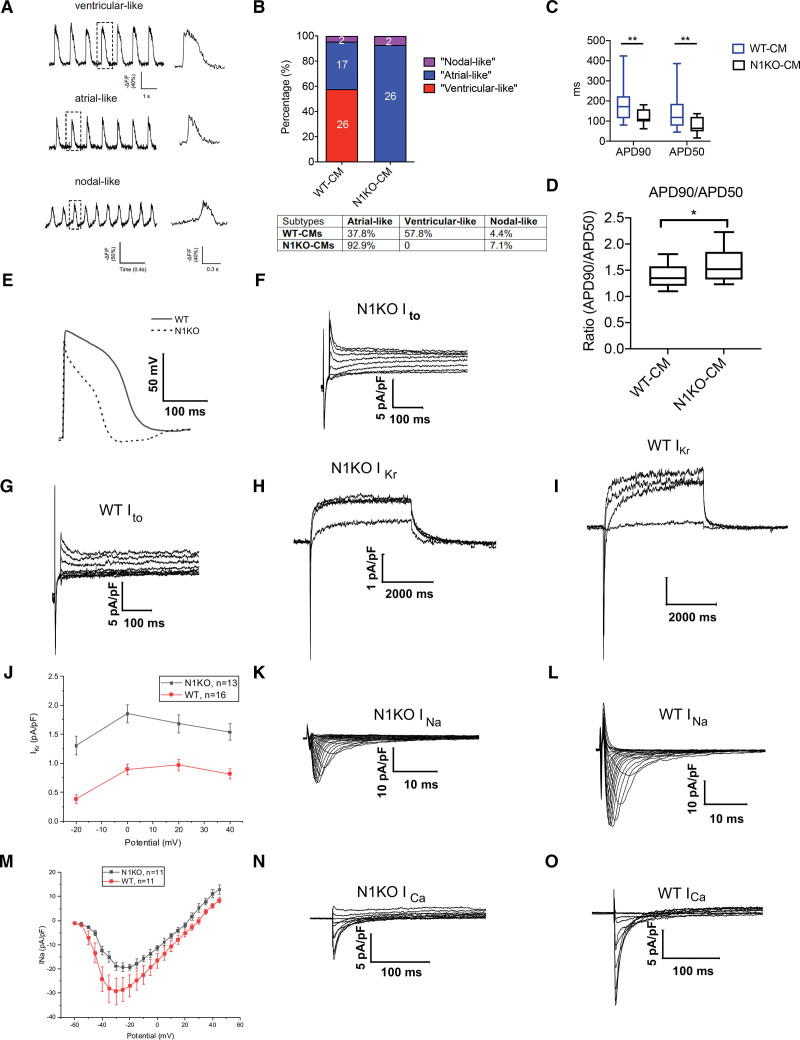
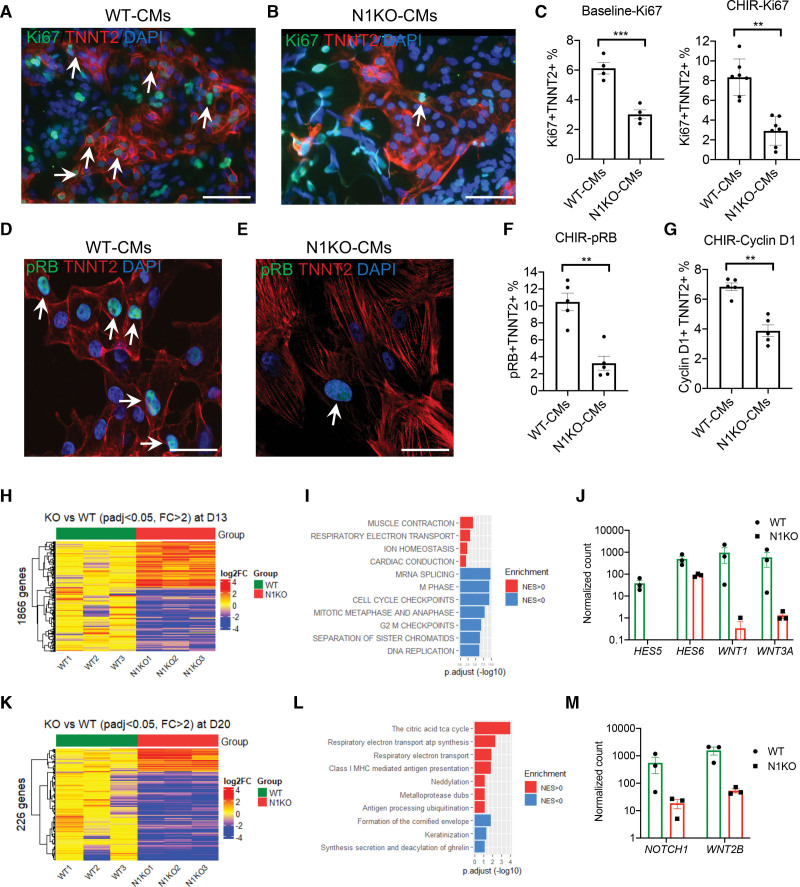
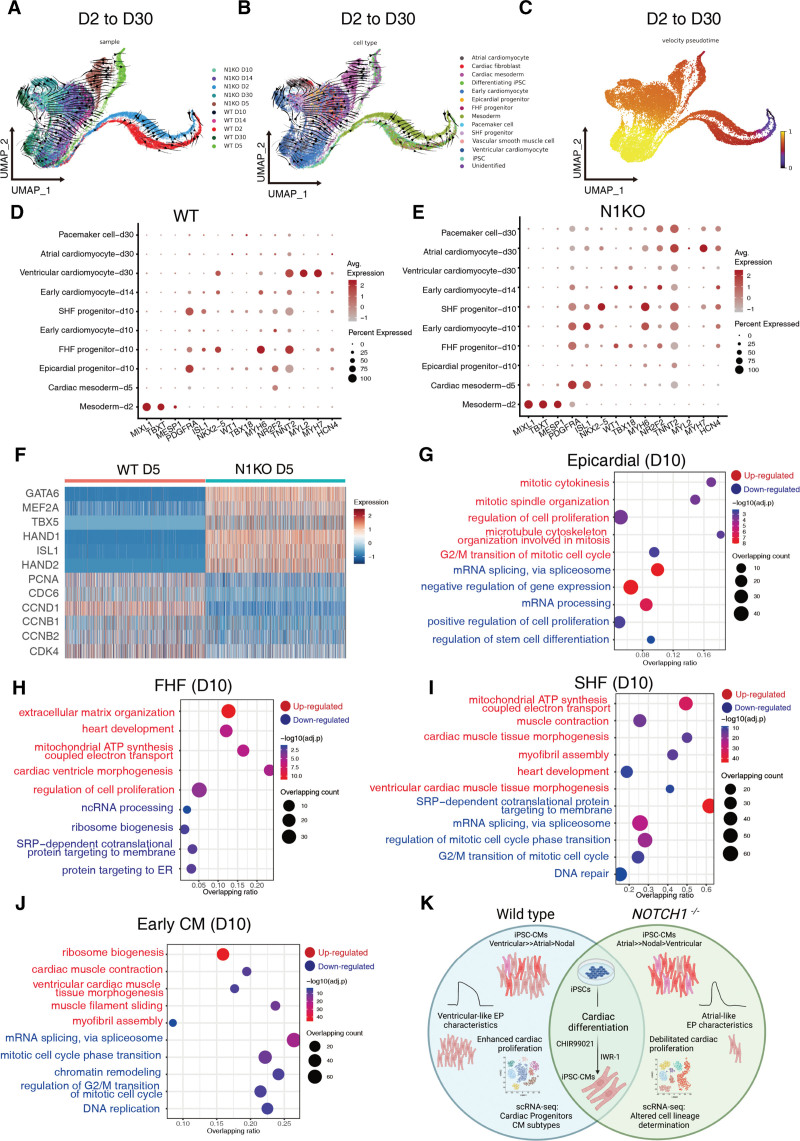
Results: Human NOTCH1 knockout induced pluripotent stem cells are able to generate functional cardiomyocytes and endothelial cells, suggesting that NOTCH1 is not required for mesoderm differentiation and cardiovascular development in vitro. However, disruption of NOTCH1 blocks human ventricular-like cardiomyocyte differentiation but promotes atrial-like cardiomyocyte generation through shortening the action potential duration. NOTCH1 deficiency leads to defective proliferation of early human cardiomyocytes, and transcriptomic analysis indicates that pathways involved in cell cycle progression and mitosis are downregulated in NOTCH1 knockout cardiomyocytes. Single-cell transcriptomic analysis reveals abnormal cell lineage determination of cardiac mesoderm, which is manifested by the biased differentiation toward epicardial and second heart field progenitors at the expense of first heart field progenitors in NOTCH1 knockout cell populations.
Conclusions: NOTCH1 is essential for human ventricular-like cardiomyocyte differentiation and proliferation through balancing cell fate determination of cardiac mesoderm and modulating cell cycle progression. Because first heart field progenitors primarily contribute to the left ventricle, we speculate that pathogenic NOTCH1 variants lead to biased differentiation of first heart field progenitors, blocked ventricular-like cardiomyocyte differentiation, and defective cardiomyocyte proliferation, which collaboratively contribute to left ventricular hypoplasia in hypoplastic left heart syndrome.
Keywords: NOTCH1; cardiac lineage differentiation; cardiomyocyte proliferation; hypoplastic left heart syndrome; induced pluripotent stem cells; mesoderm; single-cell RNA-seq.
Figures
Comment in
-
hiPSCs as a Unique Platform to Model Cardiogenesis in NOTCH1-Associated HLHS: hiPSCs to Model Complex Congenital Heart Defects.Circ Res. 2023 Jan 20;132(2):205-207. doi: 10.1161/CIRCRESAHA.122.322353. Epub 2023 Jan 19. Circ Res. 2023. PMID: 36656965 No abstract available.
References
-
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009 - PubMed
-
- Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97:1235–1294. doi: 10.1152/physrev.00005.2017 - PubMed
-
- High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. doi: 10.1038/nrg2279 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
