

Gene regulatory network reconfiguration in direct lineage reprogramming
- PMID: 36584685
- PMCID: PMC9860067
- DOI: 10.1016/j.stemcr.2022.11.010
Gene regulatory network reconfiguration in direct lineage reprogramming
Abstract
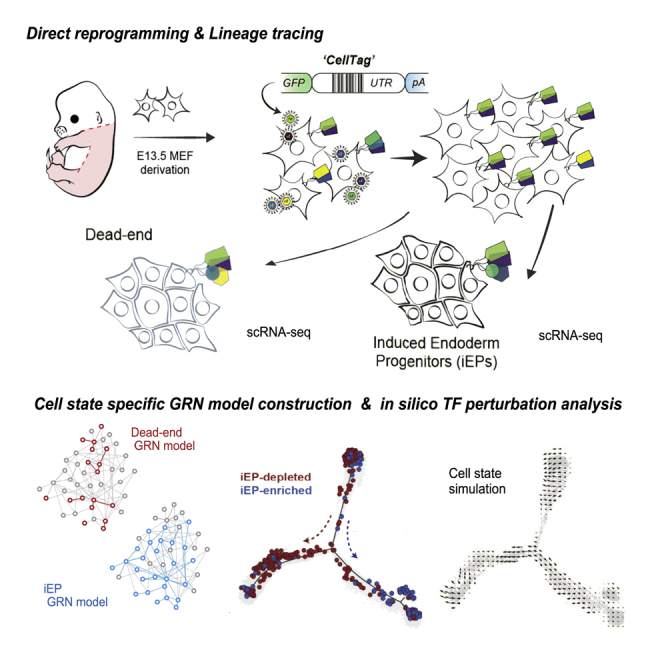
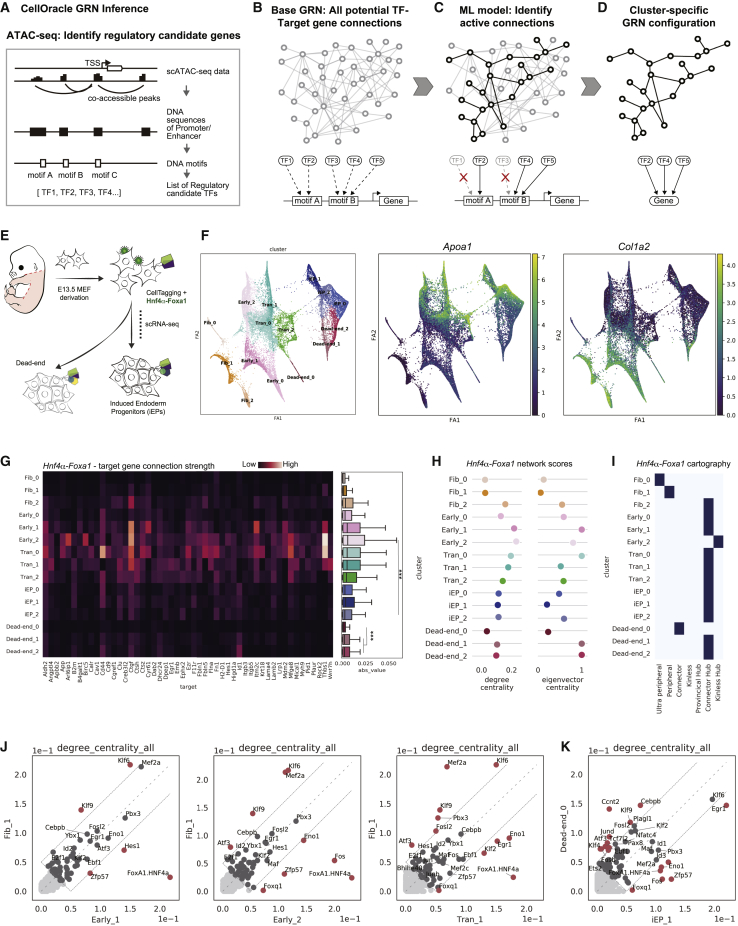
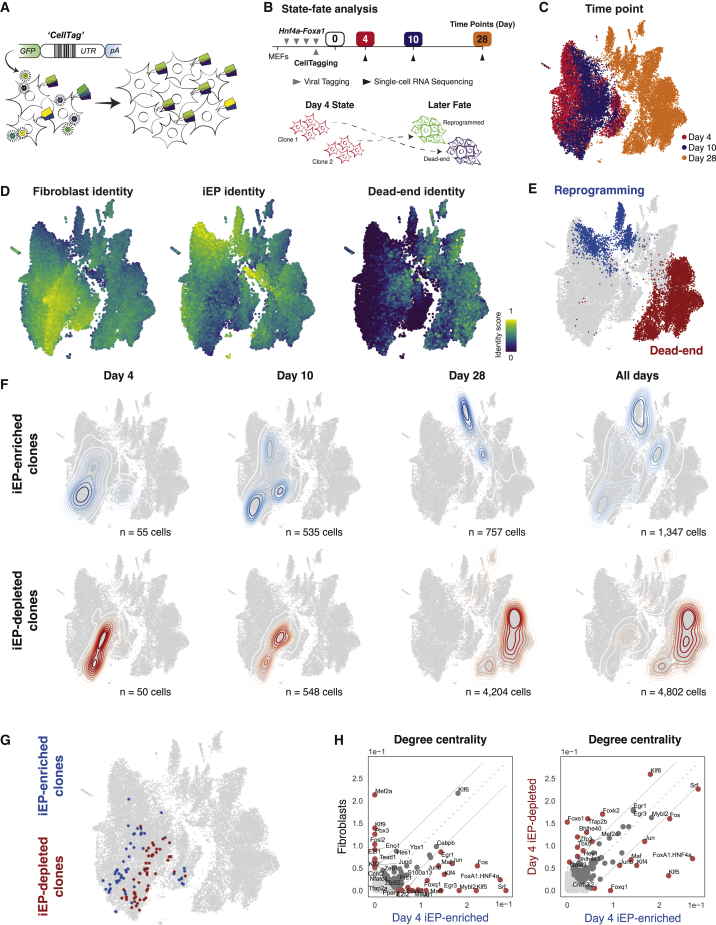
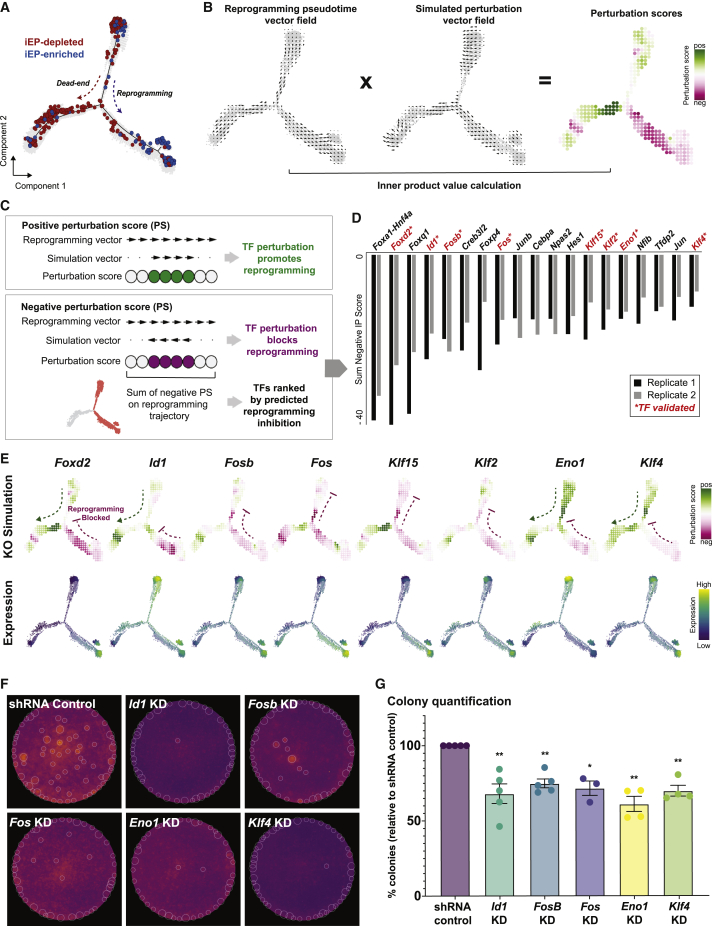
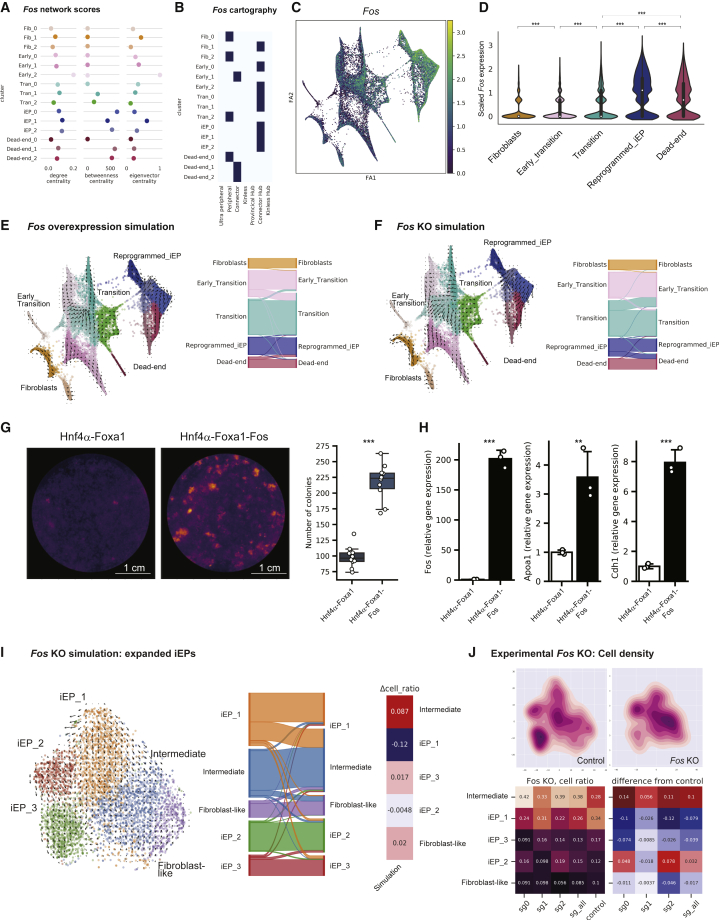
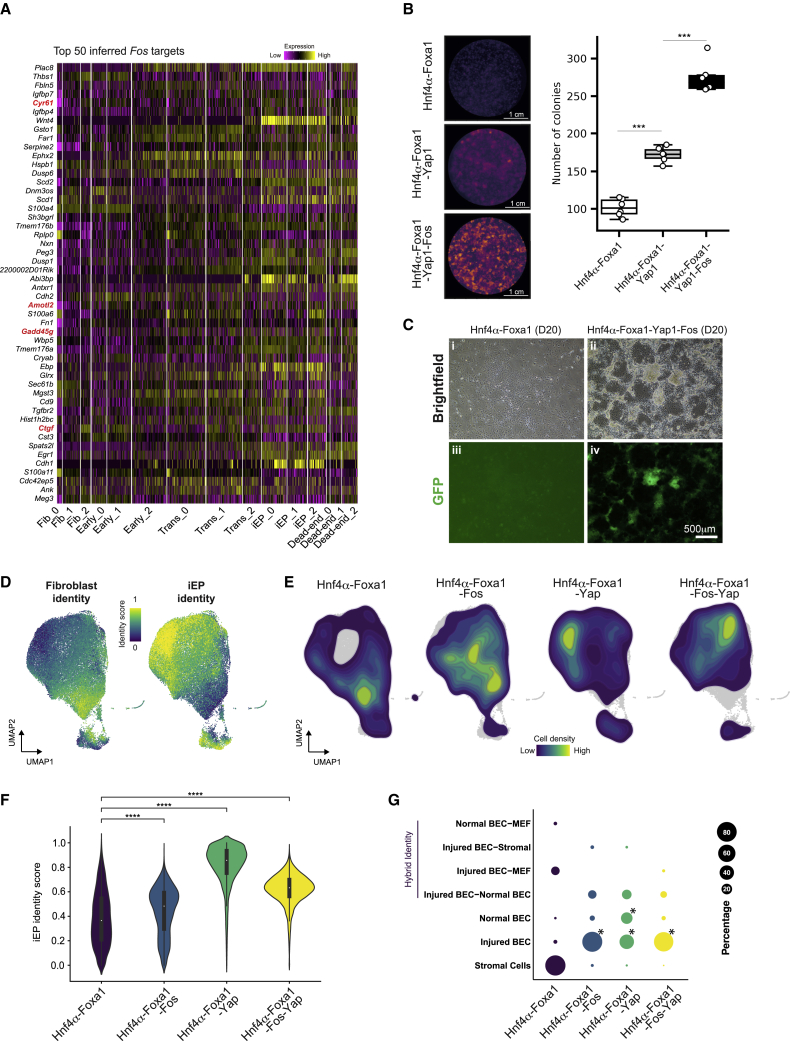
In direct lineage conversion, transcription factor (TF) overexpression reconfigures gene regulatory networks (GRNs) to reprogram cell identity. We previously developed CellOracle, a computational method to infer GRNs from single-cell transcriptome and epigenome data. Using inferred GRNs, CellOracle simulates gene expression changes in response to TF perturbation, enabling in silico interrogation of network reconfiguration. Here, we combine CellOracle analysis with lineage tracing of fibroblast to induced endoderm progenitor (iEP) conversion, a prototypical direct reprogramming paradigm. By linking early network state to reprogramming outcome, we reveal distinct network configurations underlying successful and failed fate conversion. Via in silico simulation of TF perturbation, we identify new factors to coax cells into successfully converting their identity, uncovering a central role for the AP-1 subunit Fos with the Hippo signaling effector, Yap1. Together, these results demonstrate the efficacy of CellOracle to infer and interpret cell-type-specific GRN configurations, providing new mechanistic insights into lineage reprogramming.
Keywords: cell fate prediction; direct lineage reprogramming; gene perturbation simulation; gene regulatory networks; machine learning; single-cell analysis.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interests S.A.M. is a co-founder of CapyBio LLC.
Figures
References
-
- Adamson B., Norman T.M., Jost M., Cho M.Y., Nuñez J.K., Chen Y., Villalta J.E., Gilbert L.A., Horlbeck M.A., Hein M.Y., et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867–1882.e21. doi: 10.1016/j.cell.2016.11.048. - DOI - PMC - PubMed
-
- Bocchi V.D., Conforti P., Vezzoli E., Besusso D., Cappadona C., Lischetti T., Galimberti M., Ranzani V., Bonnal R.J.P., De Simone M., et al. The coding and long noncoding single-cell atlas of the developing human fetal striatum. Science. 2021;372:eabf5759. doi: 10.1126/science.abf5759. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
