

The clinical utility of integrative genomics in childhood cancer extends beyond targetable mutations
- PMID: 36585449
- PMCID: PMC9970873
- DOI: 10.1038/s43018-022-00474-y
The clinical utility of integrative genomics in childhood cancer extends beyond targetable mutations
Abstract
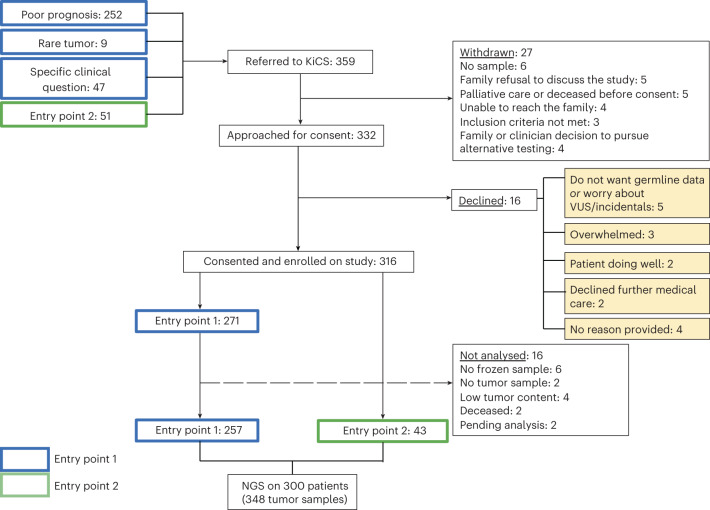
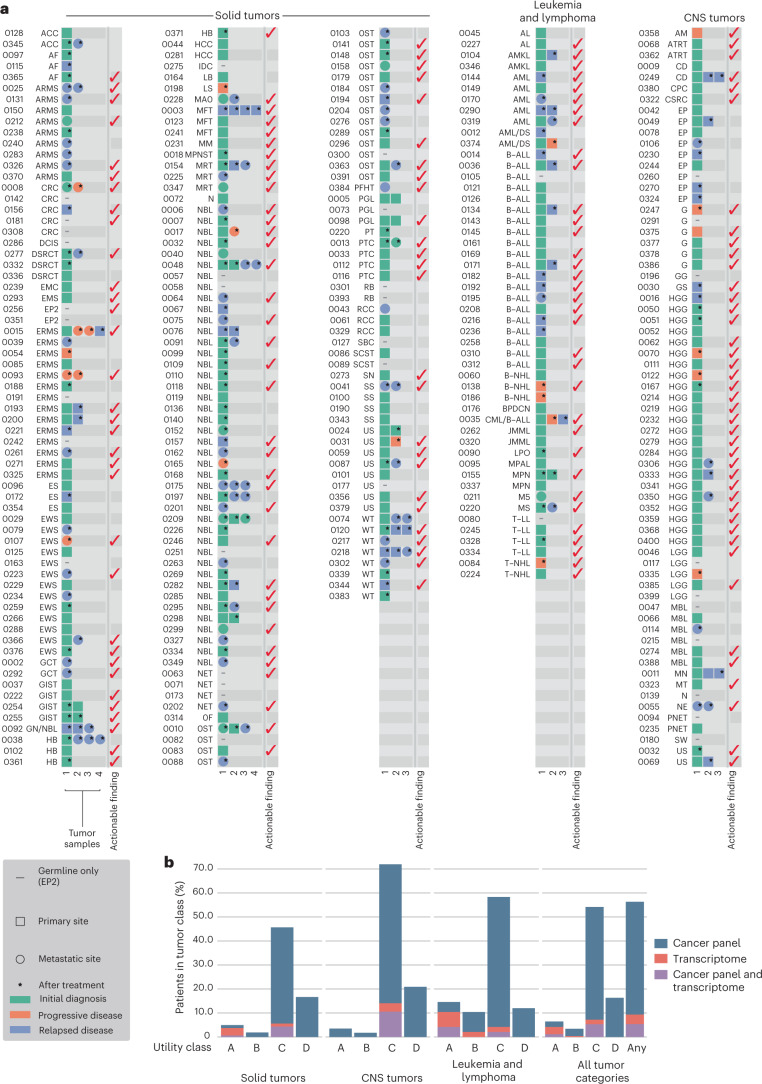
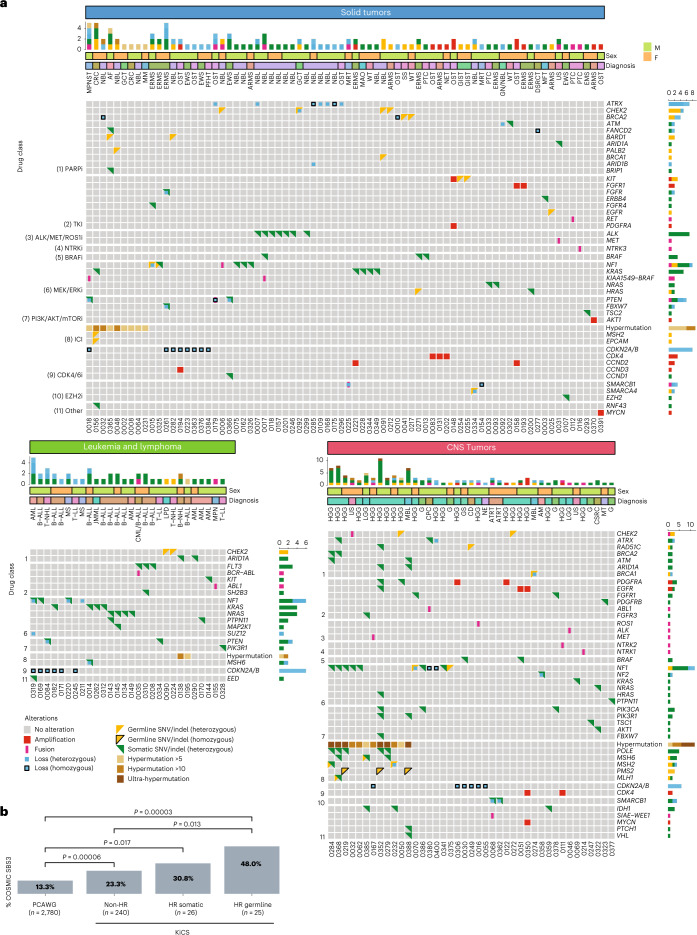
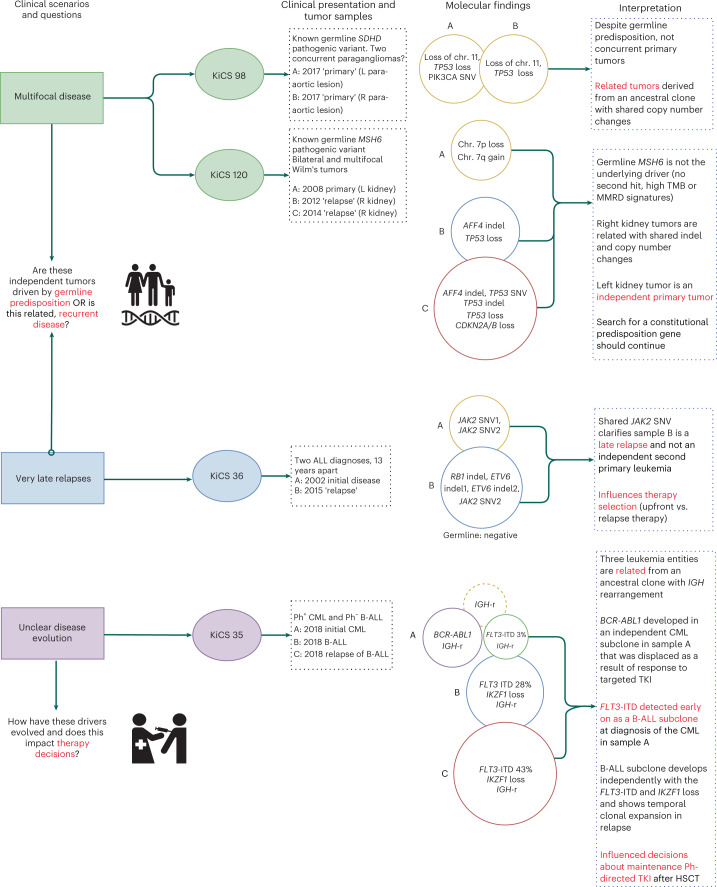
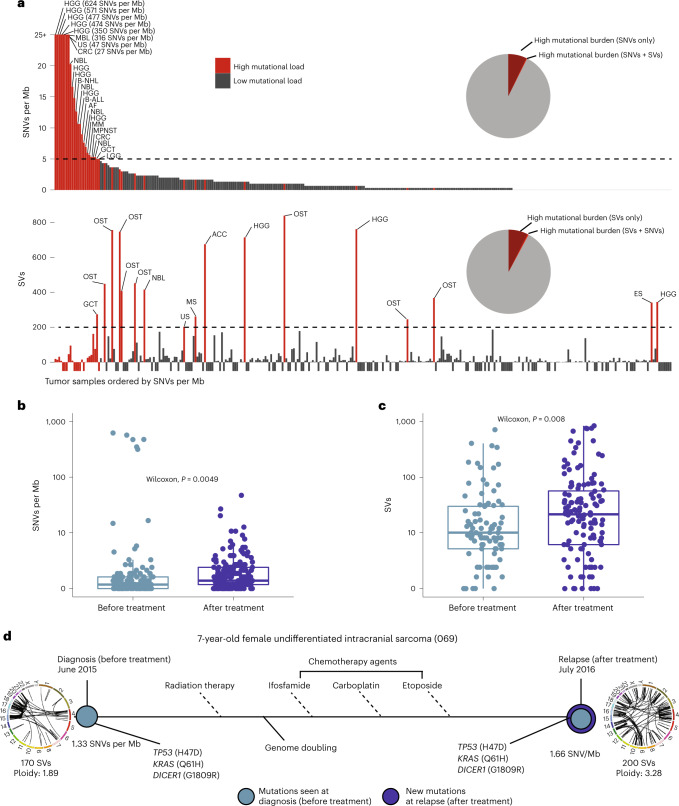
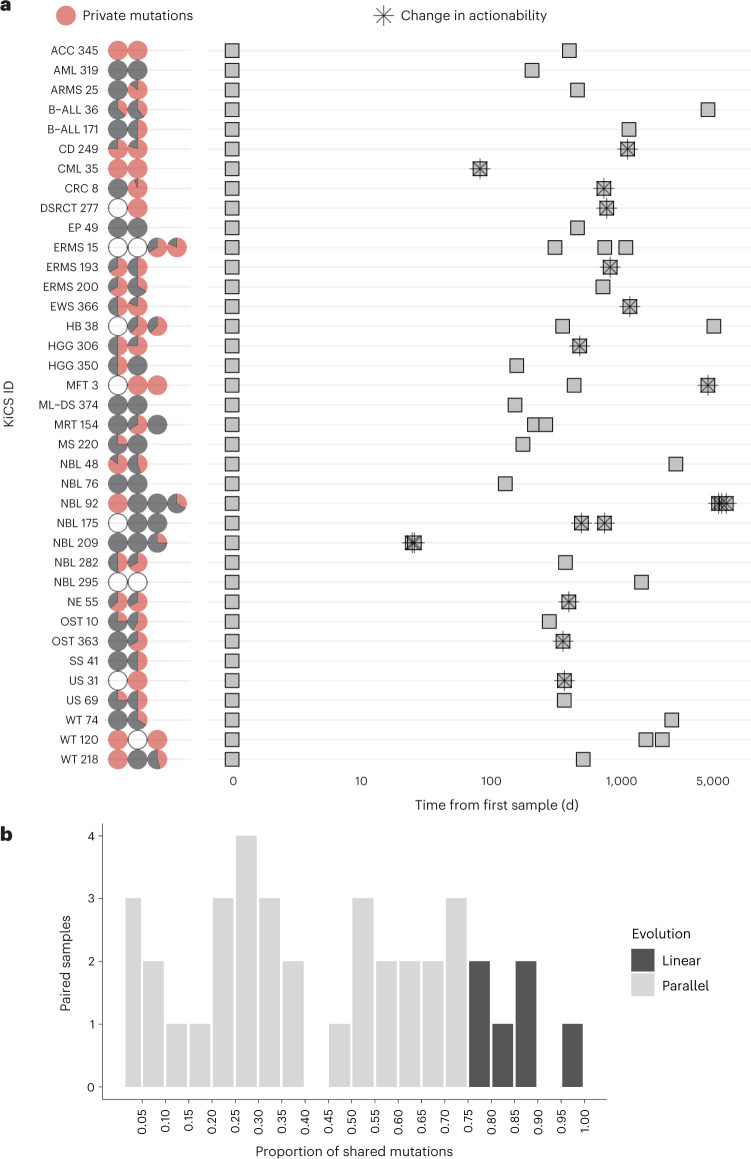
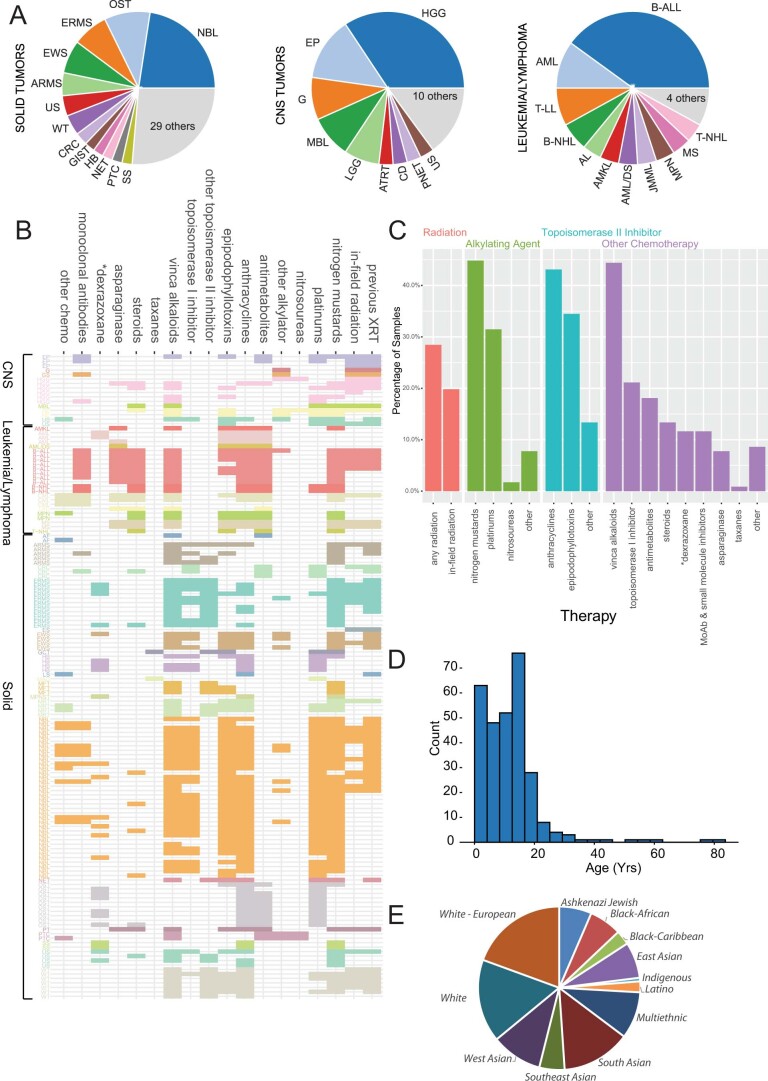
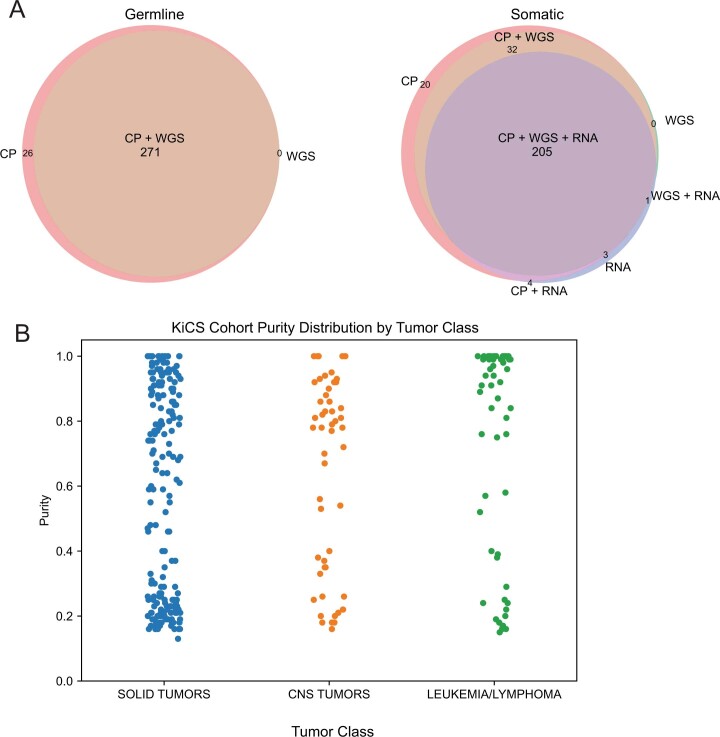
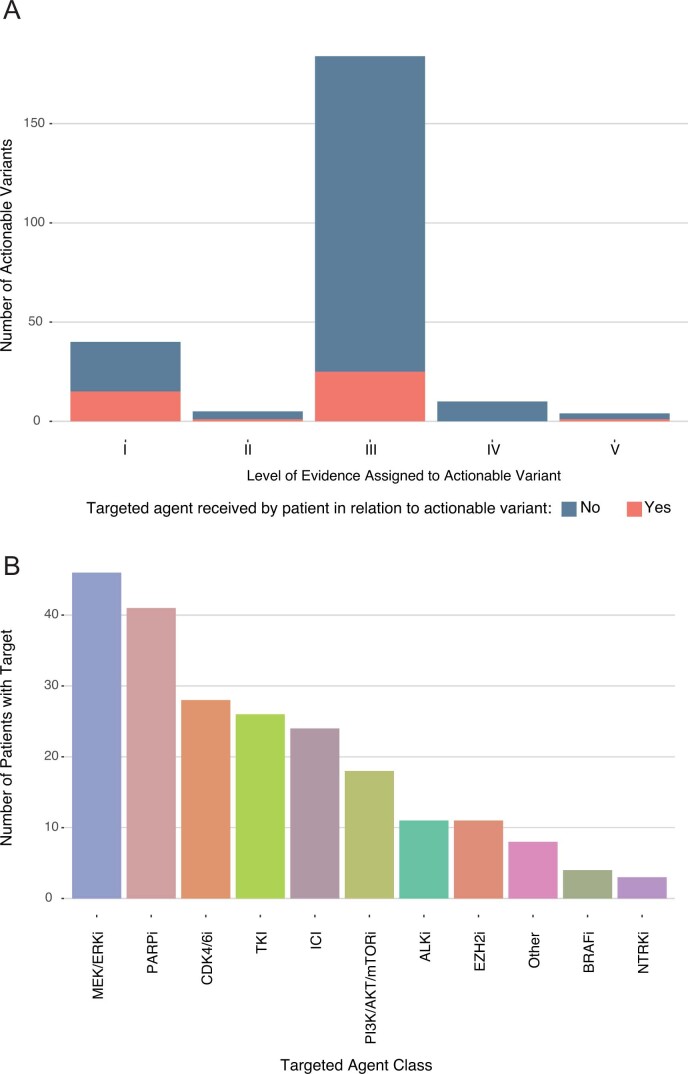
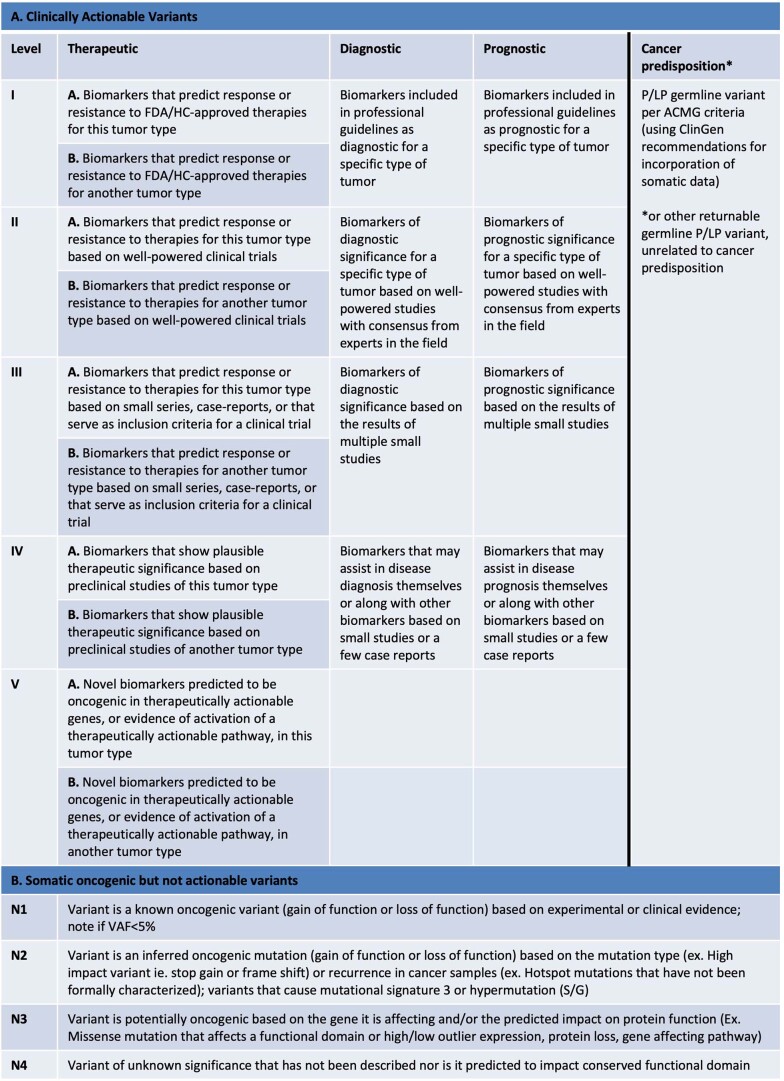
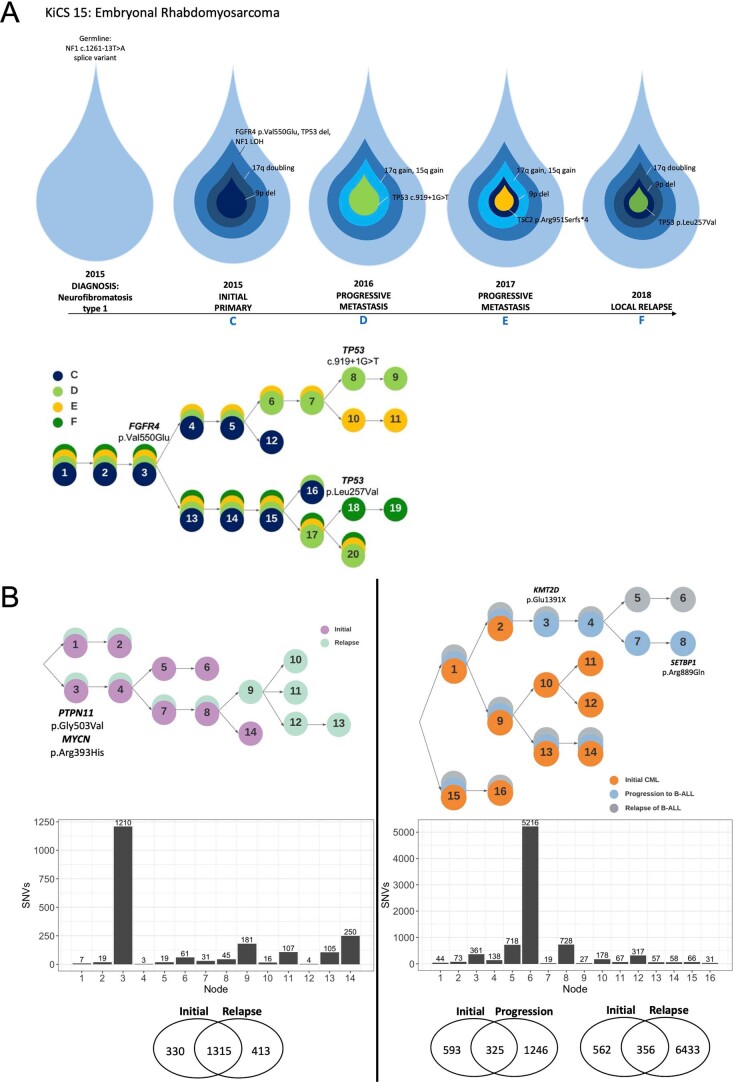
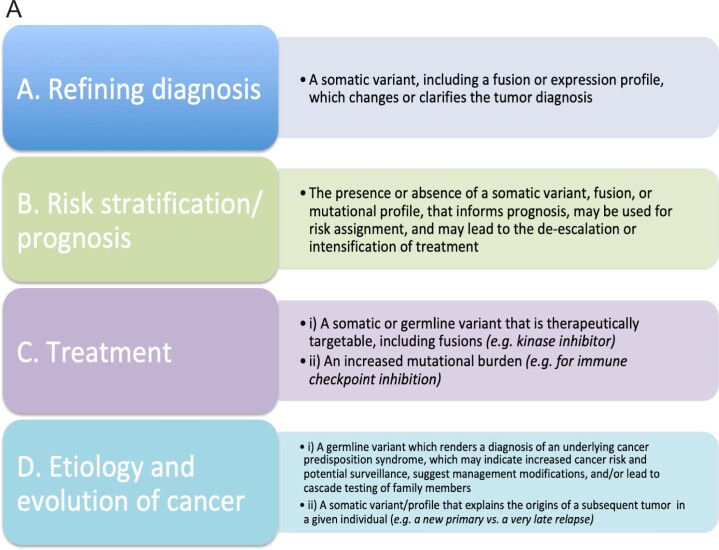
We conducted integrative somatic-germline analyses by deeply sequencing 864 cancer-associated genes, complete genomes and transcriptomes for 300 mostly previously treated children and adolescents/young adults with cancer of poor prognosis or with rare tumors enrolled in the SickKids Cancer Sequencing (KiCS) program. Clinically actionable variants were identified in 56% of patients. Improved diagnostic accuracy led to modified management in a subset. Therapeutically targetable variants (54% of patients) were of unanticipated timing and type, with over 20% derived from the germline. Corroborating mutational signatures (SBS3/BRCAness) in patients with germline homologous recombination defects demonstrates the potential utility of PARP inhibitors. Mutational burden was significantly elevated in 9% of patients. Sequential sampling identified changes in therapeutically targetable drivers in over one-third of patients, suggesting benefit from rebiopsy for genomic analysis at the time of relapse. Comprehensive cancer genomic profiling is useful at multiple points in the care trajectory for children and adolescents/young adults with cancer, supporting its integration into early clinical management.
© 2022. The Author(s).
Conflict of interest statement
D.A.M.: consultancy/advisory board for ymAbs Therapeutics, EUSA Pharma and Clarity Pharmaceuticals. A.S and F.C.: a patent application has been filed on an RNA-seq-based tumor classifier algorithm.
Figures

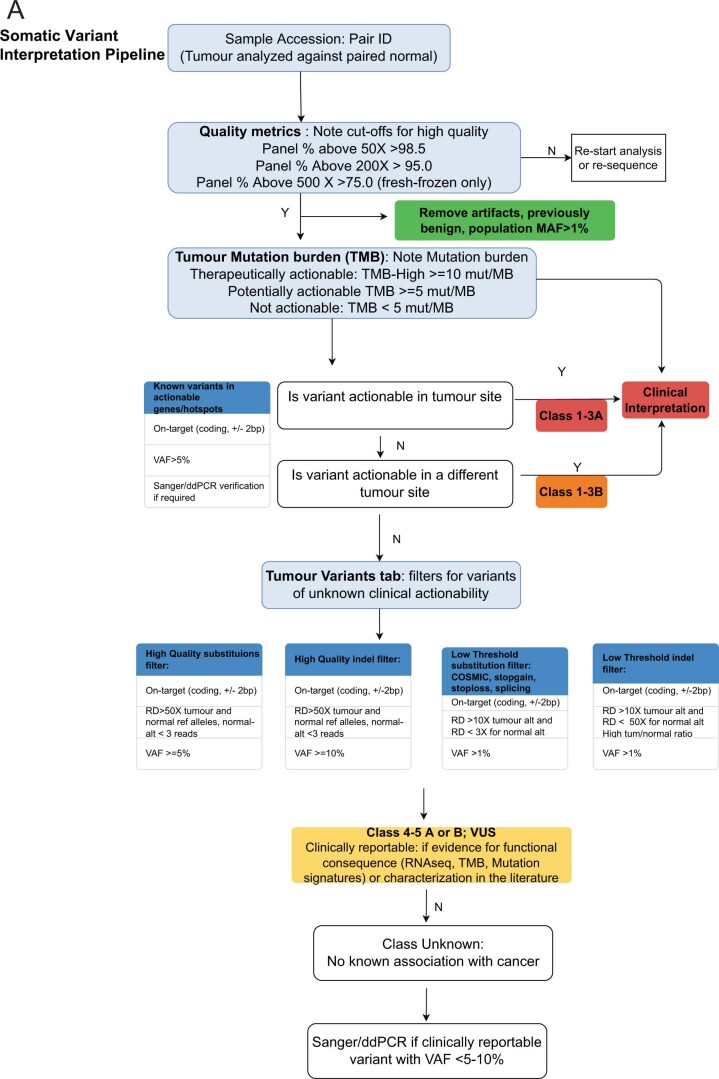
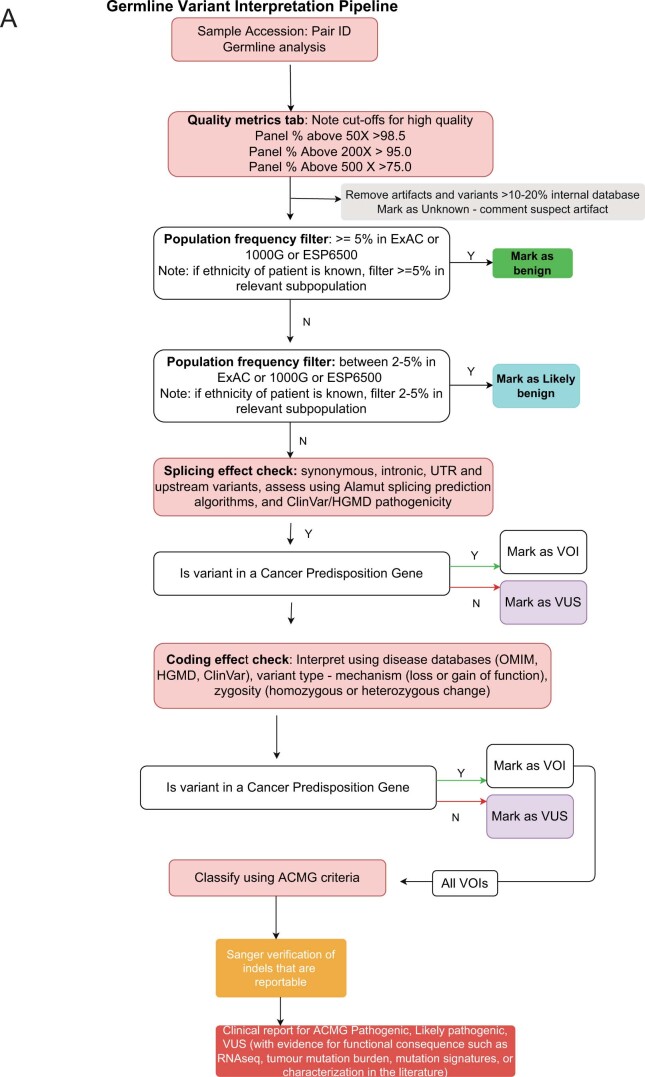
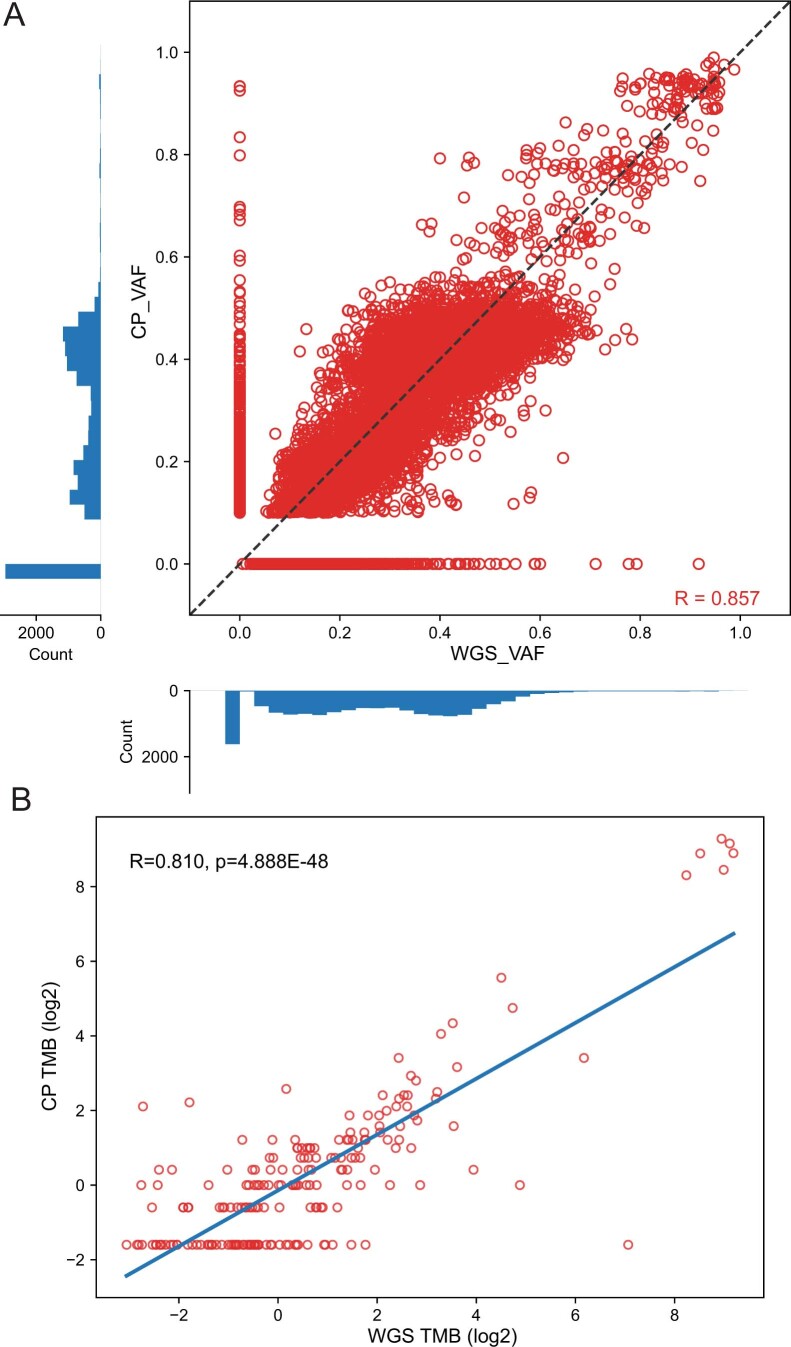
Comment in
-
Enhancing childhood cancer targetability.Nat Cancer. 2023 Feb;4(2):153-155. doi: 10.1038/s43018-022-00472-0. Nat Cancer. 2023. PMID: 36585448 No abstract available.
References
-
- Grobner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018); erratum 559, E10 (2018). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
