

Multiomics in primary and metastatic breast tumors from the AURORA US network finds microenvironment and epigenetic drivers of metastasis
- PMID: 36585450
- PMCID: PMC9886551
- DOI: 10.1038/s43018-022-00491-x
Multiomics in primary and metastatic breast tumors from the AURORA US network finds microenvironment and epigenetic drivers of metastasis
Abstract
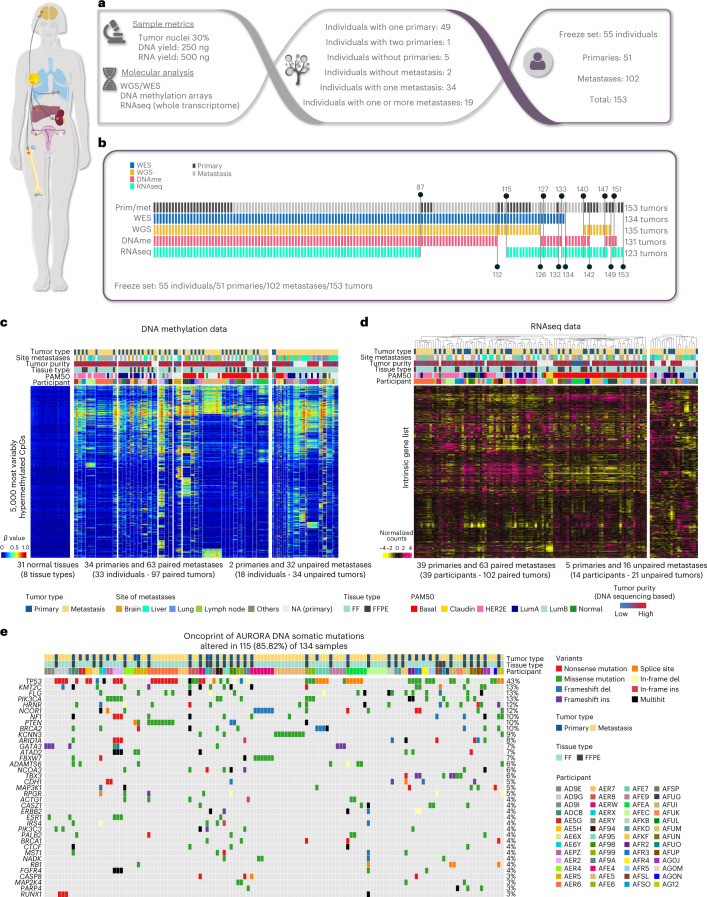
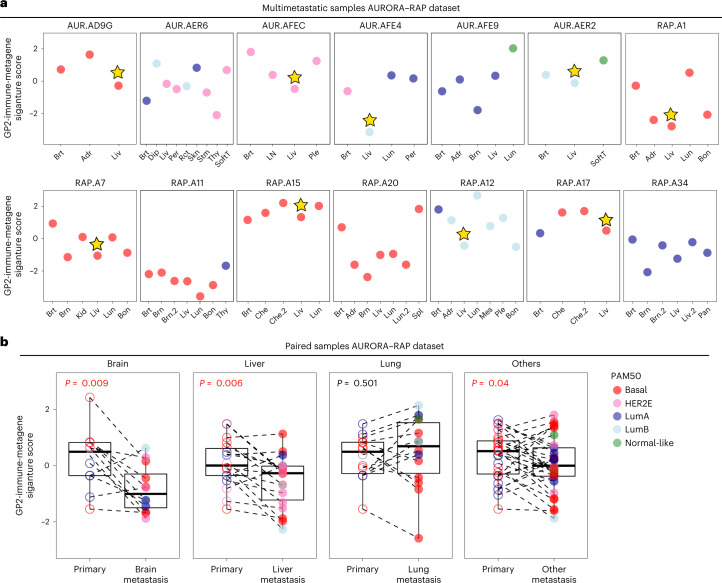
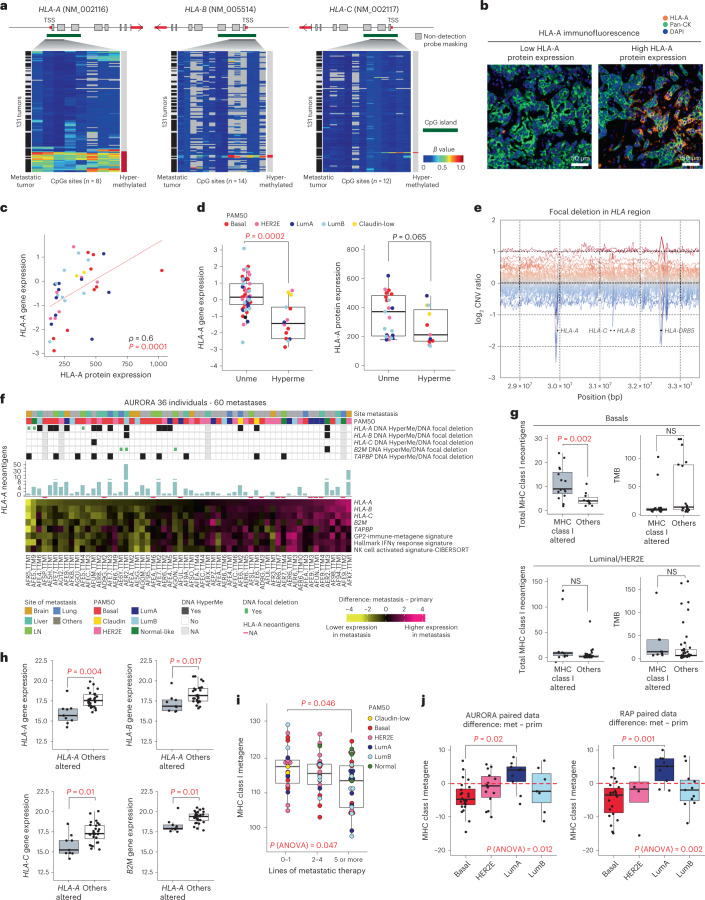
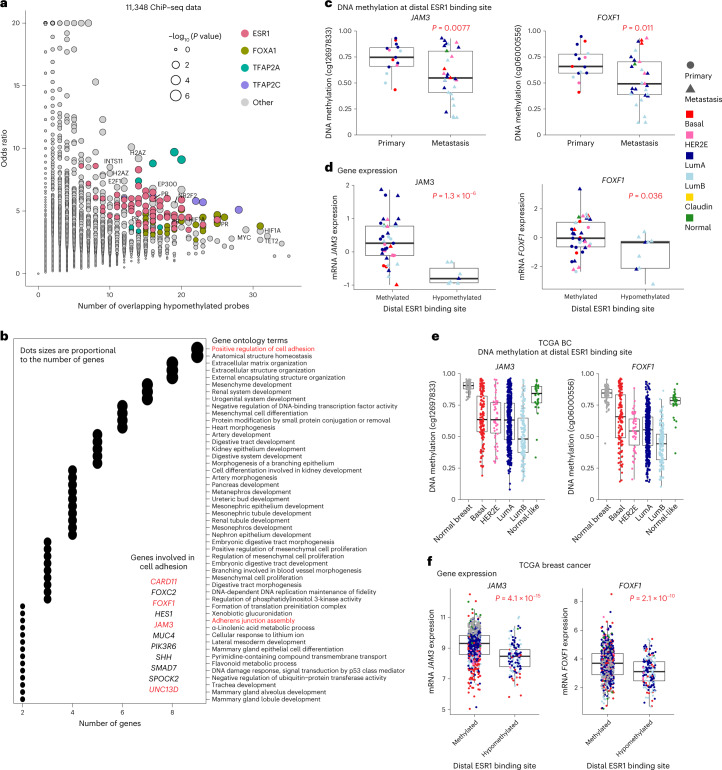
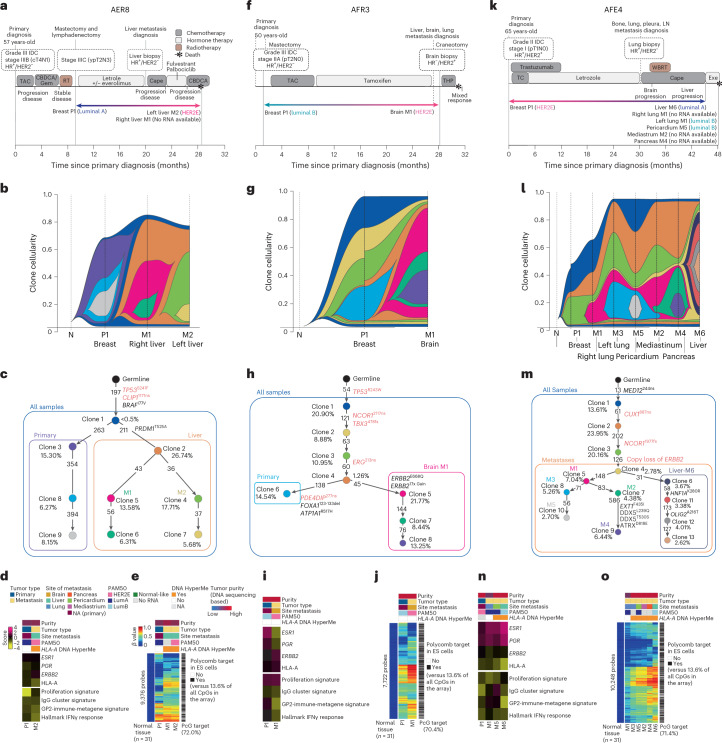
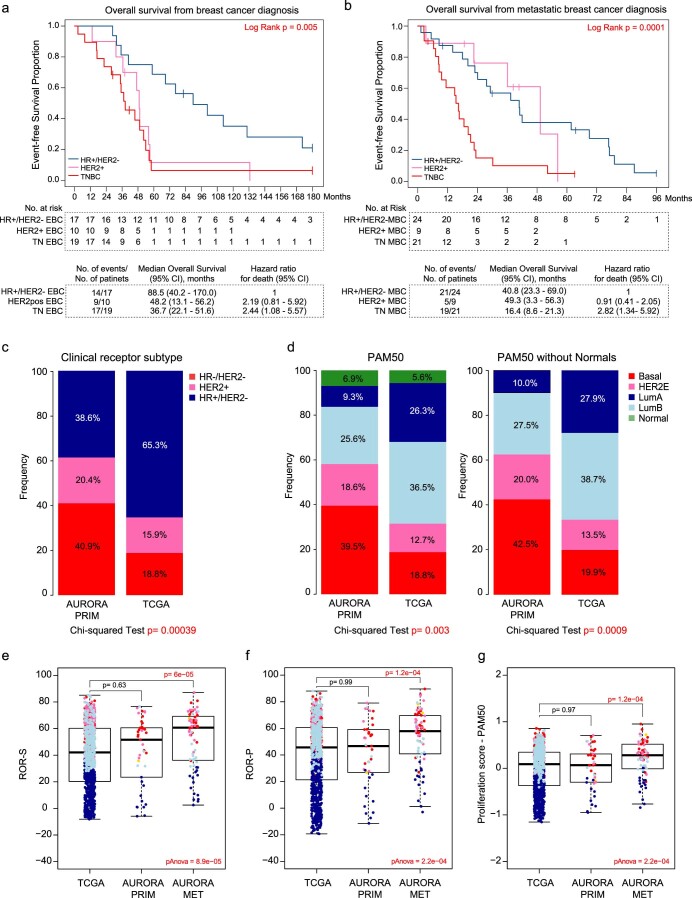
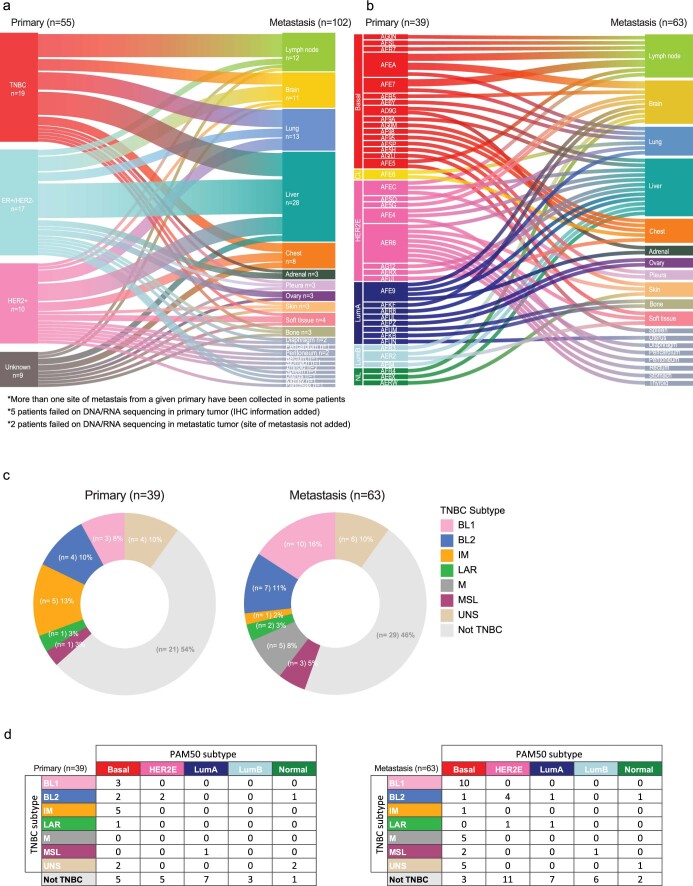
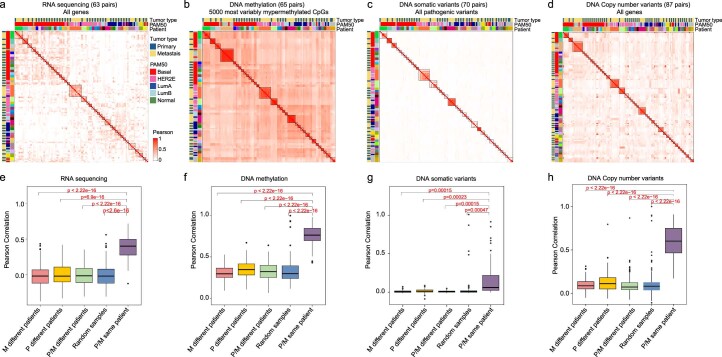
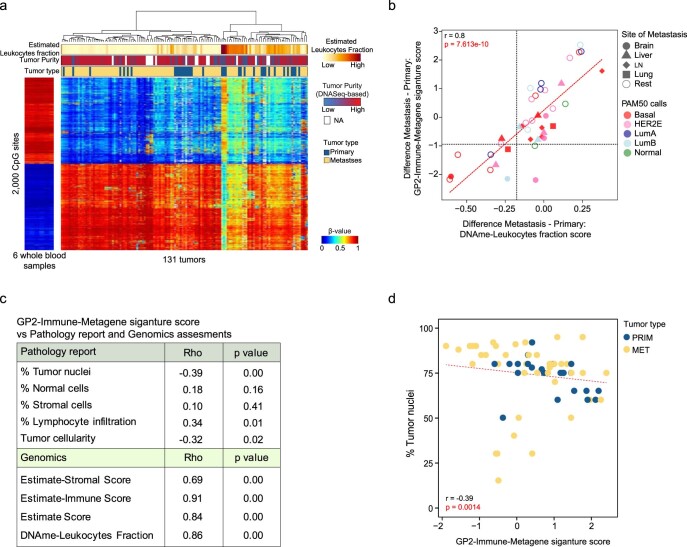
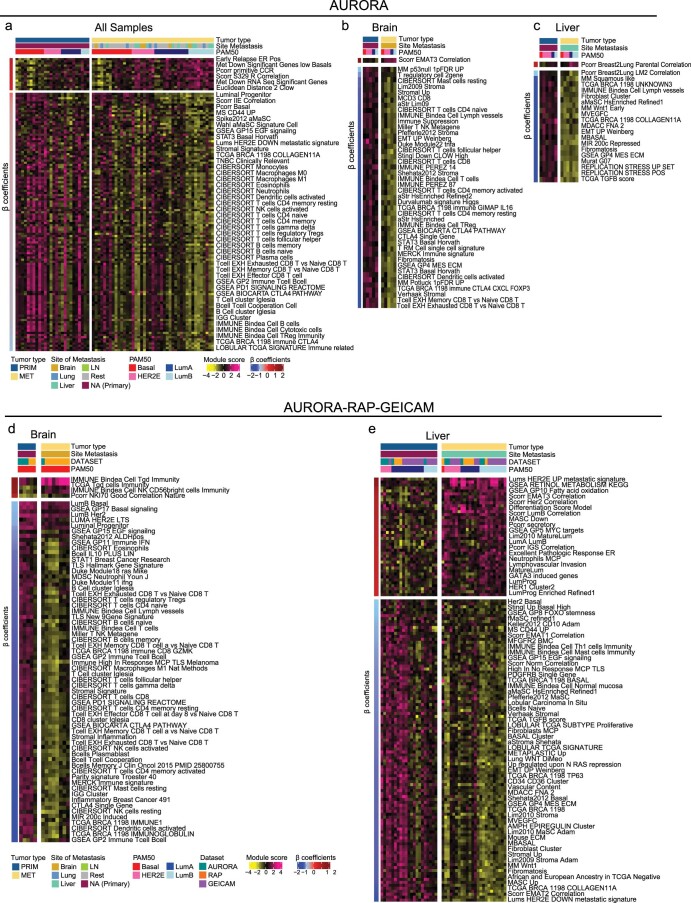
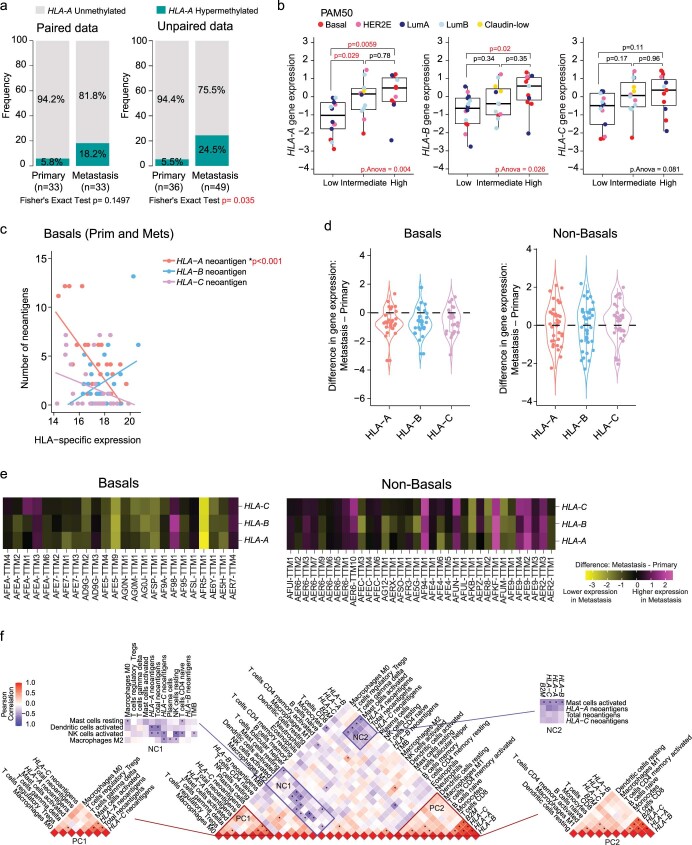
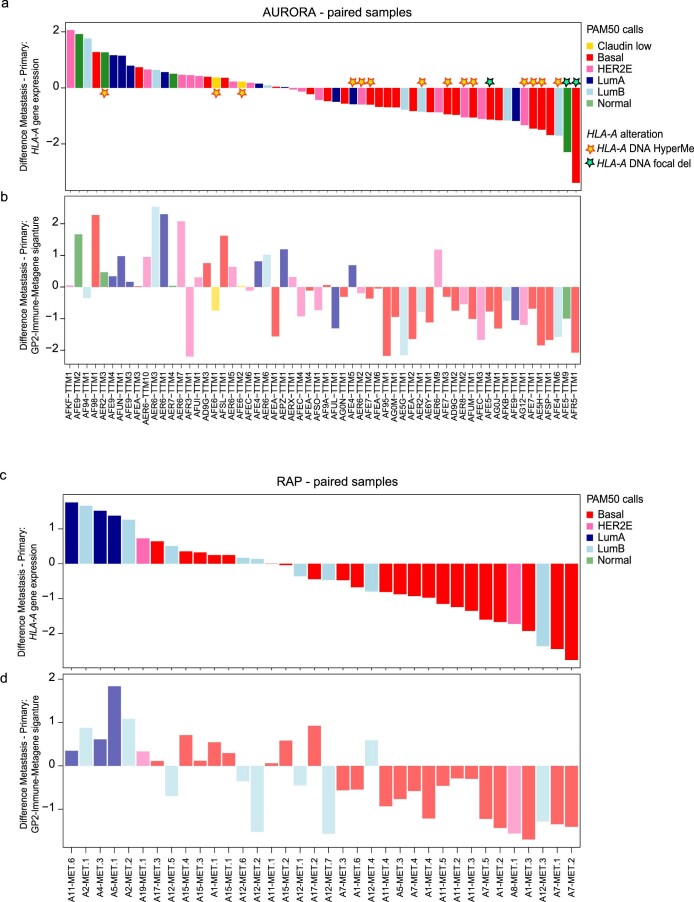
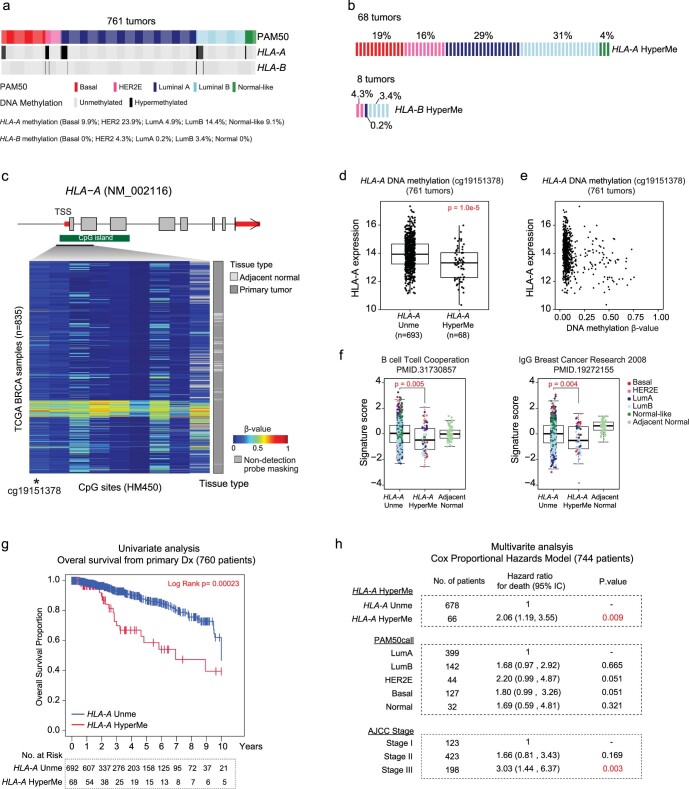
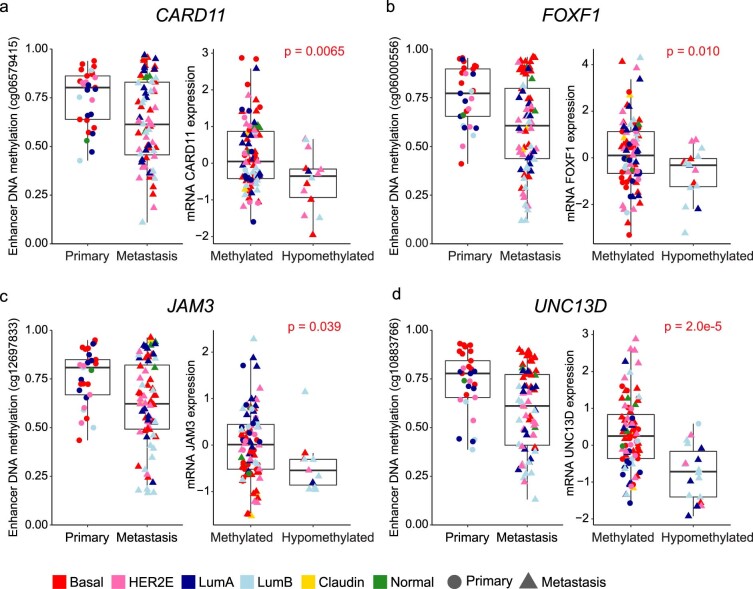
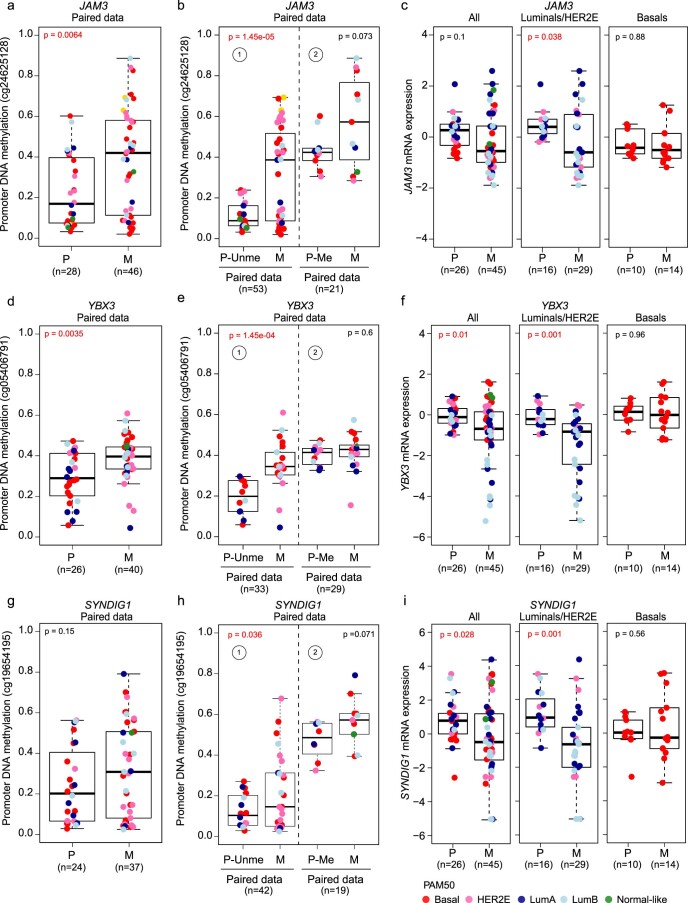
The AURORA US Metastasis Project was established with the goal to identify molecular features associated with metastasis. We assayed 55 females with metastatic breast cancer (51 primary cancers and 102 metastases) by RNA sequencing, tumor/germline DNA exome and low-pass whole-genome sequencing and global DNA methylation microarrays. Expression subtype changes were observed in ~30% of samples and were coincident with DNA clonality shifts, especially involving HER2. Downregulation of estrogen receptor (ER)-mediated cell-cell adhesion genes through DNA methylation mechanisms was observed in metastases. Microenvironment differences varied according to tumor subtype; the ER+/luminal subtype had lower fibroblast and endothelial content, while triple-negative breast cancer/basal metastases showed a decrease in B and T cells. In 17% of metastases, DNA hypermethylation and/or focal deletions were identified near HLA-A and were associated with reduced expression and lower immune cell infiltrates, especially in brain and liver metastases. These findings could have implications for treating individuals with metastatic breast cancer with immune- and HER2-targeting therapies.
© 2022. The Author(s).
Conflict of interest statement
The following authors disclose conflicts of interest. C.M.P. is listed as an inventor on patent applications on the Breast PAM50 assay and is an equity stock holder and consultant of BioClassifier LLC. J.S.P. is listed as an inventor on patent applications on the Breast PAM50 assay. H.S. has authorship and equity in AnchorDX, authorship with Illumina and an IP license with TruDiagnostics, Inc. B.H.P. has royalties: Horizon Discovery, Ltd.; Consultant: EQRx, Sermonix, Hologics, Jackson Laboratories, Guardant Health Inc; Unpaid consultant: Tempus; Consultant and ownership interest: Celcuity; Research Contracts: GE Healthcare, Lilly and Pfizer. I.K. has: Consulting Fees (e.g. advisory boards); Author; Bristol Meyers Squibb, Daiichi/Sankyo, Macrogenics, Context Therapeutics, Taiho Oncology, Genentech/Roche, Seattle Genetics. Contracted Research; Author; Genentech/Roche, Pfizer. Other; Author; Novartis, Merck (DSMB member). C.A. has: Research funding: Puma, Lilly, Merck, Seattle Genetics, Nektar, Tesaro, G1 Therapeutics, ZION, Novartis, Pfizer; Compensated consultant role: Genentech, Eisai, IPSEN, Seattle Genetics, AstraZeneca, Novartis, Immunomedics, Elucida, Athenex; Royalties: UpToDate, Jones and Bartlet. M.F.R. has: Consulting Fees (e.g. advisory boards); Author; Macrogenics, Daiichi, and Genentech. Contracted Research; Author; Pfizer. R.N. is an author with Aduro, AstraZeneca, Athenex, Celgene, Daiichi Sankyo, Inc., Genentech, MacroGenics, Merck, Novartis, Pfizer, Puma, Syndax. Contracted Research; Author; AstraZeneca, Celgene, Concept Therapeutics, Genentech/Roche, Immunomedics, Merck, Odonate Therapeutics, Pfizer, Seattle Genetics. Other; Author; DSMB:G1 Therapeutics; Steering Committee: OBI Pharm, Inc. N.U.L. has: Consulting Fees (e.g. advisory boards); Author; Seattle genetics, Puma and Daichii. Contracted Research; Author; Genentech, Seattle Genetics, Pfizer. C.I. has Consulting Fees (e.g. advisory boards); Author; Pfizer, AstraZeneca, Genentech, Novartis, Puma, Seattle Genetics, Sanofi, Eisai, Biotheranostics, and Gilead. Royalties; Author Wolters Kluwer (UpToDate), McGraw Hill (Goodman and Gilman’s); Research funding (to institution) Merck, Seattle Genetics, Pfizer, GlaxoSmithKline. M.C.L. has: Author; Eisai, Genentech, GRAIL, Janssen, Merck, Novartis, Seattle Genetics, Tesaro. J.M.B. has: Receipt of Intellectual Property Rights / Patent Holder; Author; Provisional patents regarding immunotherapy targets and biomarkers in cancer. Consulting Fees (e.g. advisory boards); Author; Novartis. Contracted Research; Author; Genentech/Roche, Bristol Myers Squibb, and Incyte Corporation. P.W.L. has: Consulting Fees (e.g. advisory boards); Progenity, Inc., Stock Options; Author; AnchorDx, Author: Progenity, Inc.Illumina, Inc., IP License; TruDiagnostic Inc. A.C.G.-C.: Research funding (to Institution) from Merck, Gilead Sciences, and AstraZeneca. All remaining authors have no relevant disclosures.
Figures

References
-
- Aftimos P, et al. Genomic and transcriptomic analyses of breast cancer primaries and matched metastases in AURORA, the Breast International Group (BIG) molecular screening initiative. Cancer Discov. 2021;11:2796–2811. doi: 10.1158/2159-8290.CD-20-1647. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
