

Targeting peroxisomal fatty acid oxidation improves hepatic steatosis and insulin resistance in obese mice
- PMID: 36586435
- PMCID: PMC9898756
- DOI: 10.1016/j.jbc.2022.102845
Targeting peroxisomal fatty acid oxidation improves hepatic steatosis and insulin resistance in obese mice
Abstract
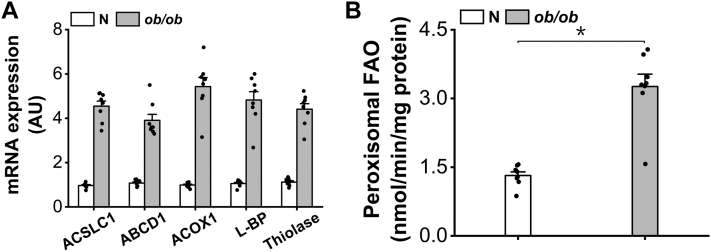
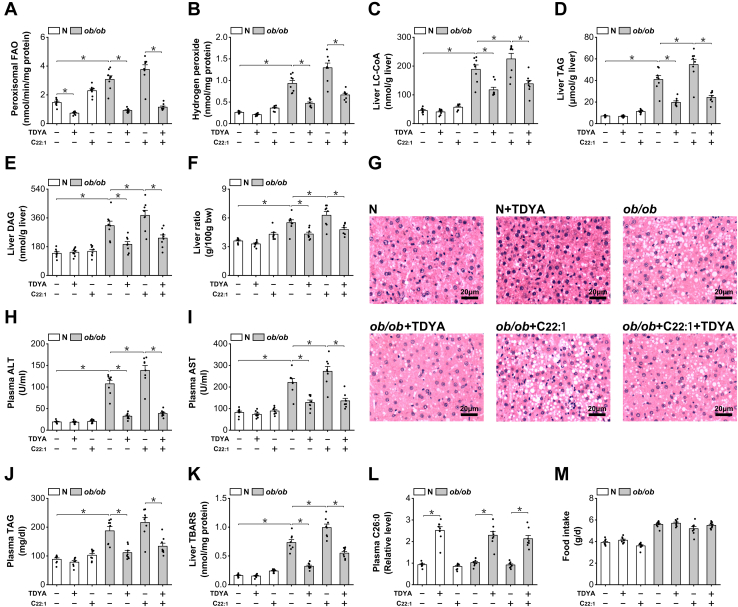
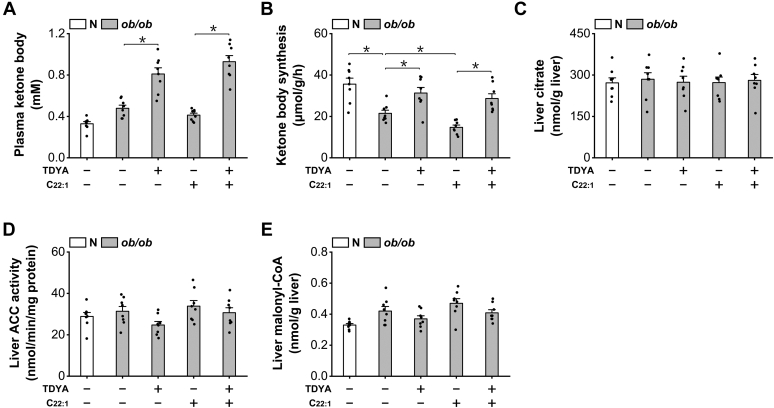
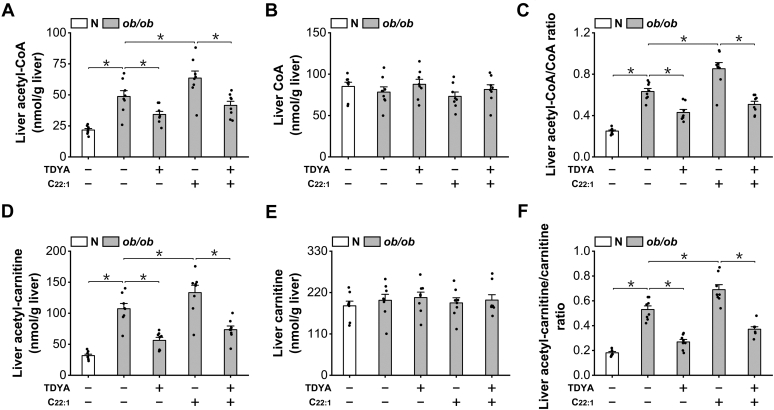
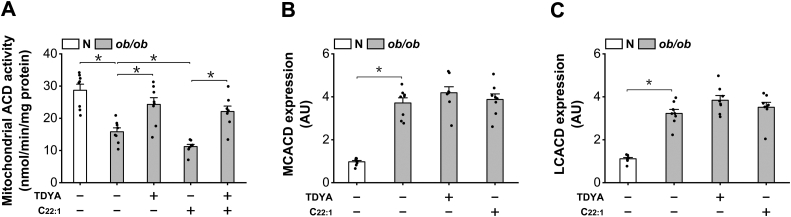
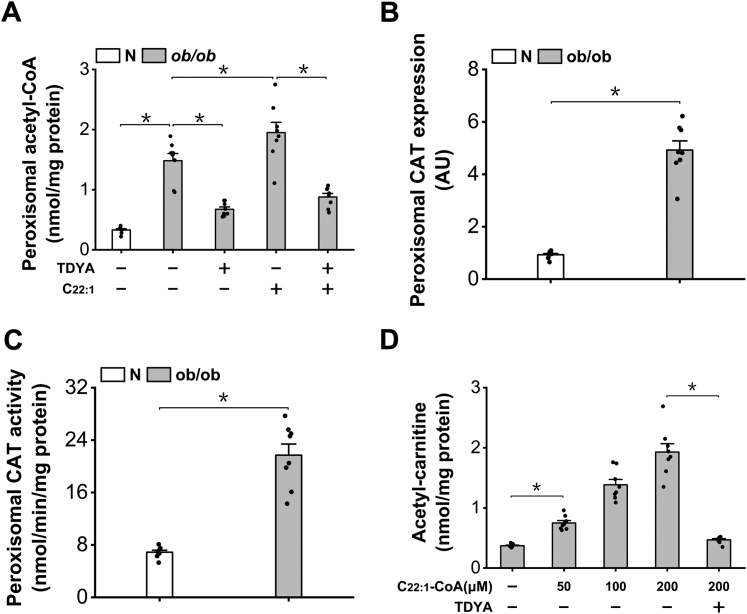
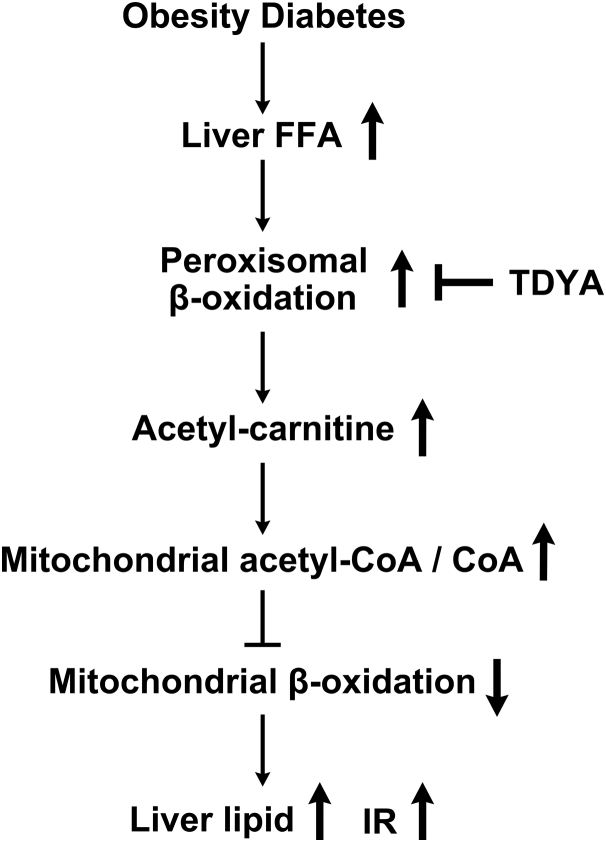
Obesity and diabetes normally cause mitochondrial dysfunction and hepatic lipid accumulation, while fatty acid synthesis is suppressed and malonyl-CoA is depleted in the liver of severe obese or diabetic animals. Therefore, a negative regulatory mechanism might work for the control of mitochondrial fatty acid metabolism that is independent of malonyl-CoA in the diabetic animals. As mitochondrial β-oxidation is controlled by the acetyl-CoA/CoA ratio, and the acetyl-CoA generated in peroxisomal β-oxidation could be transported into mitochondria via carnitine shuttles, we hypothesize that peroxisomal β-oxidation might play a role in regulating mitochondrial fatty acid oxidation and inducing hepatic steatosis under the condition of obesity or diabetes. This study reveals a novel mechanism by which peroxisomal β-oxidation controls mitochondrial fatty acid oxidation in diabetic animals. We determined that excessive oxidation of fatty acids by peroxisomes generates considerable acetyl-carnitine in the liver of diabetic mice, which significantly elevates the mitochondrial acetyl-CoA/CoA ratio and causes feedback suppression of mitochondrial β-oxidation. Additionally, we found that specific suppression of peroxisomal β-oxidation enhances mitochondrial fatty acid oxidation by reducing acetyl-carnitine formation in the liver of obese mice. In conclusion, we suggest that induction of peroxisomal fatty acid oxidation serves as a mechanism for diabetes-induced hepatic lipid accumulation. Targeting peroxisomal β-oxidation might be a promising pathway in improving hepatic steatosis and insulin resistance as induced by obesity or diabetes.
Keywords: acetyl-carnitine; fatty acid oxidation; mitochondria; obesity; peroxisomes.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures

References
-
- Brady R.O., Gurin S. Biosynthesis of labeled fatty acids and cholesterol in experimental diabetes. J. Bio. Chem. 1950;187:589–596. - PubMed
-
- McGarry J.D., Stark M.J., Foster D.W. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J. Biol. Chem. 1978;274:2766–2772. - PubMed
-
- Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 2008;28:253–272. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
