

Probing conformational landscapes and mechanisms of allosteric communication in the functional states of the ABL kinase domain using multiscale simulations and network-based mutational profiling of allosteric residue potentials
- PMID: 36586979
- PMCID: PMC11184971
- DOI: 10.1063/5.0133826
Probing conformational landscapes and mechanisms of allosteric communication in the functional states of the ABL kinase domain using multiscale simulations and network-based mutational profiling of allosteric residue potentials
Abstract
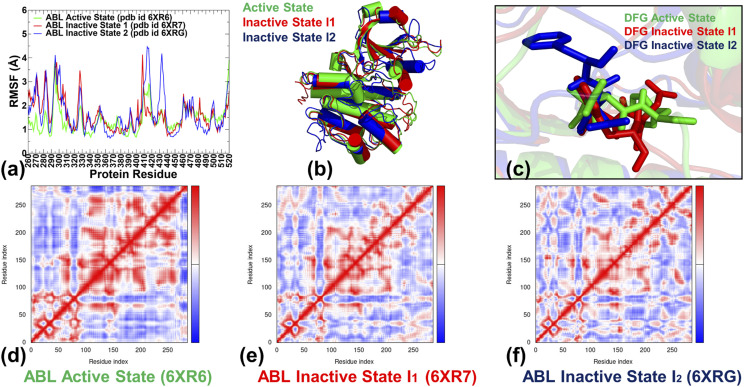
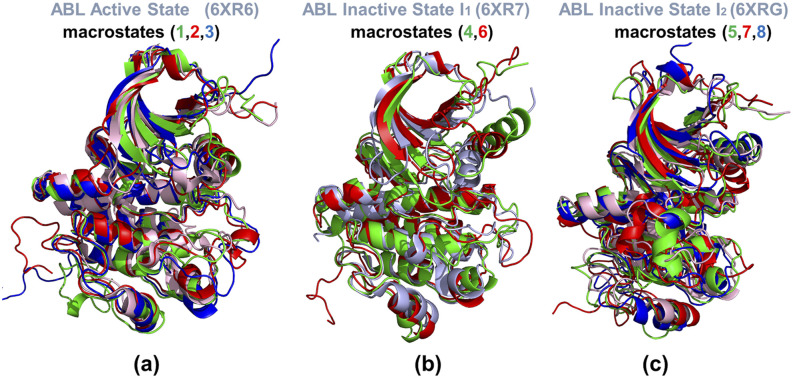
In the current study, multiscale simulation approaches and dynamic network methods are employed to examine the dynamic and energetic details of conformational landscapes and allosteric interactions in the ABL kinase domain that determine the kinase functions. Using a plethora of synergistic computational approaches, we elucidate how conformational transitions between the active and inactive ABL states can employ allosteric regulatory switches to modulate intramolecular communication networks between the ATP site, the substrate binding region, and the allosteric binding pocket. A perturbation-based network approach that implements mutational profiling of allosteric residue propensities and communications in the ABL states is proposed. Consistent with biophysical experiments, the results reveal functionally significant shifts of the allosteric interaction networks in which preferential communication paths between the ATP binding site and substrate regions in the active ABL state become suppressed in the closed inactive ABL form, which in turn features favorable allosteric coupling between the ATP site and the allosteric binding pocket. By integrating the results of atomistic simulations with dimensionality reduction methods and Markov state models, we analyze the mechanistic role of macrostates and characterize kinetic transitions between the ABL conformational states. Using network-based mutational scanning of allosteric residue propensities, this study provides a comprehensive computational analysis of long-range communications in the ABL kinase domain and identifies conserved regulatory hotspots that modulate kinase activity and allosteric crosstalk between the allosteric pocket, ATP binding site, and substrate binding regions.
Conflict of interest statement
The authors have no conflicts to disclose.
Figures







References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous