

Co-Occurring Atypical Galactosemia and Wilson Disease
- PMID: 36588756
- PMCID: PMC9801321
- DOI: 10.1159/000524004
Co-Occurring Atypical Galactosemia and Wilson Disease
Abstract
Introduction: Classic galactosemia is a disorder of the galactose metabolism and is inherited as an autosomal recessive disease. It is caused by a complete or severe deficiency of galactose-1-phosphate uridyltransferase (GALT), and in rare cases, atypical galactosemia can manifest at older ages. Wilson disease (WD) is a disorder of the copper metabolism that, like galactosemia, is inherited as an autosomal recessive disease. Hepatic, neurological, or psychiatric symptoms can be seen, independently or in combination, and symptoms vary from family to family. We present here a patient diagnosed with both WD and galactosemia.
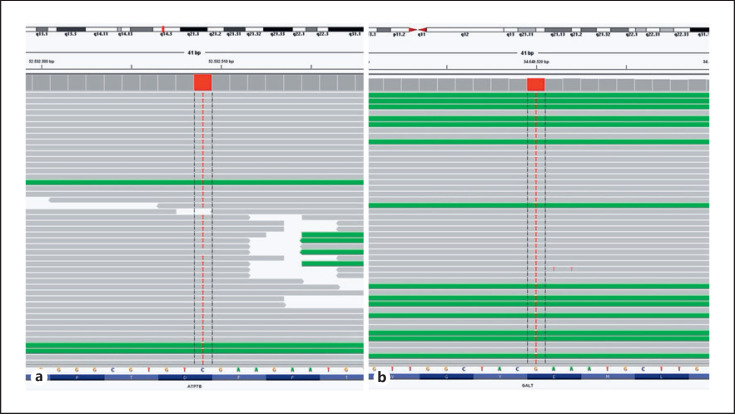
Case presentation: A 6-year-old girl was referred to our center with elevated transaminase levels and hepatosplenomegaly. The child, birthweight of 2,200 g, was born to first-degree consanguineous parents after a full-term uneventful pregnancy and was hospitalized in the neonatal period due to indirect hyperbilirubinemia, gastrointestinal bleeding, diarrhea lasting 2 weeks, and elevated liver enzymes. Hepatosplenomegaly was evident at the time of admission, a cataract was detected, and a neuropsychiatric evaluation revealed borderline mental capacity, as well as cognitive and speech retardation. Metabolic investigations revealed no specific findings other than trace positivity of reducing substances in the urine. A liver biopsy revealed copper accumulation in hepatocytes and low ceruloplasmin levels. Although WD was suspected in the patient, this diagnosis did not explain the intellectual disability, behavioral disorder, or cataract findings. A genetic analysis revealed homozygous mutations in the ATP7B and GALT genes. The galactose-1-phosphate uridyltransferase enzyme level was found to be low, and the patient was diagnosed with coexisting WD and galactosemia.
Conclusion: Coexistences of rare genetically transmitted diseases can be seen in countries where consanguineous marriages are common (Saudi Arabia, Iran, Pakistan, etc.), as in our country, Turkey.
Keywords: Atypical manifestation; Coexistence; Expanded genetic analysis; Galactosemia; Wilson disease.
Copyright © 2022 by S. Karger AG, Basel.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Clinical and biochemical phenotypes, genotypes, and long-term outcomes of individuals with galactosemia type I from a single metabolic genetics center in Alberta.Mol Genet Metab Rep. 2024 Jan 25;38:101055. doi: 10.1016/j.ymgmr.2024.101055. eCollection 2024 Mar. Mol Genet Metab Rep. 2024. PMID: 38469090 Free PMC article.
-
Galactosemia: when is it a newborn screening emergency?Mol Genet Metab. 2012 May;106(1):7-11. doi: 10.1016/j.ymgme.2012.03.007. Epub 2012 Mar 21. Mol Genet Metab. 2012. PMID: 22483615 Review.
-
Simultaneous occurrence of various mutations and polymorphisms in cis and in trans of the galactose-1-phosphate uridyltransferase gene in a Turkish family with classical galactosemia.J Mol Med (Berl). 1998 Sep;76(10):715-9. doi: 10.1007/s001090050272. J Mol Med (Berl). 1998. PMID: 9766850
-
Unusual Presentation of Classical Galactosemia: A Case Report of Iranian Experience.Clin Case Rep. 2025 Feb 18;13(2):e70170. doi: 10.1002/ccr3.70170. eCollection 2025 Feb. Clin Case Rep. 2025. PMID: 39973892 Free PMC article.
-
The molecular biology of galactosemia.Genet Med. 1998 Nov-Dec;1(1):40-8. doi: 10.1097/00125817-199811000-00009. Genet Med. 1998. PMID: 11261429 Review.
Cited by
-
Shifting the Focus of Molecular Syndromology from Individual Diagnoses to Outcome Analyses.Mol Syndromol. 2023 Aug;14(4):267-269. doi: 10.1159/000531738. Epub 2023 Jul 18. Mol Syndromol. 2023. PMID: 37484705 Free PMC article. No abstract available.
-
Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review.Genes (Basel). 2022 Dec 16;13(12):2377. doi: 10.3390/genes13122377. Genes (Basel). 2022. PMID: 36553645 Free PMC article. Review.
-
Expanded Phenotypic Spectrum or Multiple Syndromes?Mol Syndromol. 2022 Dec;13(5):361-362. doi: 10.1159/000526893. Epub 2022 Oct 28. Mol Syndromol. 2022. PMID: 36588758 Free PMC article. No abstract available.
References
-
- Anderson S. GALT Deficiency Galactosemia. MCN Am J Matern Child Nurs. 2018;43((1)):44–51. - PubMed
-
- Cerone J, Rios A. Galactosemia. Pediatr Rev. 2019;40((Suppl 1)):24–7. - PubMed
-
- Gathof BS, Sommer M, Podskarbi T, Reichardt J, Braun A, Gresser U, et al. Characterization of two stop codon mutations in the galactose-1-phosphate uridyltransferase gene of three male galactosemic patients with severe clinical manifestation. Hum Genet. 1995;96((6)):721–5. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
