

Dissecting CD8+ T cell pathology of severe SARS-CoV-2 infection by single-cell immunoprofiling
- PMID: 36591270
- PMCID: PMC9800604
- DOI: 10.3389/fimmu.2022.1066176
Dissecting CD8+ T cell pathology of severe SARS-CoV-2 infection by single-cell immunoprofiling
Abstract
Introduction: SARS-CoV-2 infection results in varying disease severity, ranging from asymptomatic infection to severe illness. A detailed understanding of the immune response to SARS-CoV-2 is critical to unravel the causative factors underlying differences in disease severity and to develop optimal vaccines against new SARS-CoV-2 variants.
Methods: We combined single-cell RNA and T cell receptor sequencing with CITE-seq antibodies to characterize the CD8+ T cell response to SARS-CoV-2 infection at high resolution and compared responses between mild and severe COVID-19.
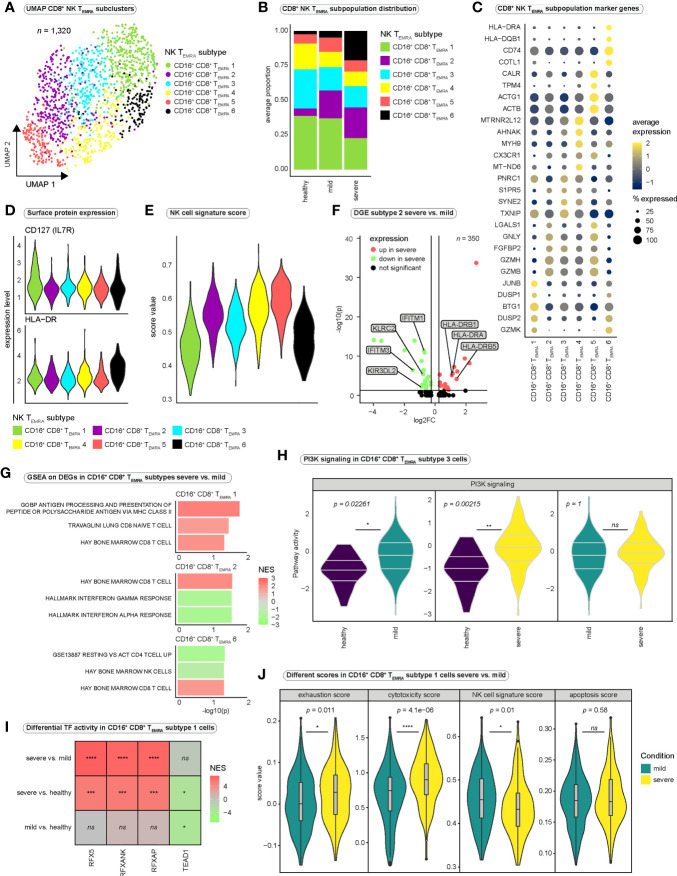
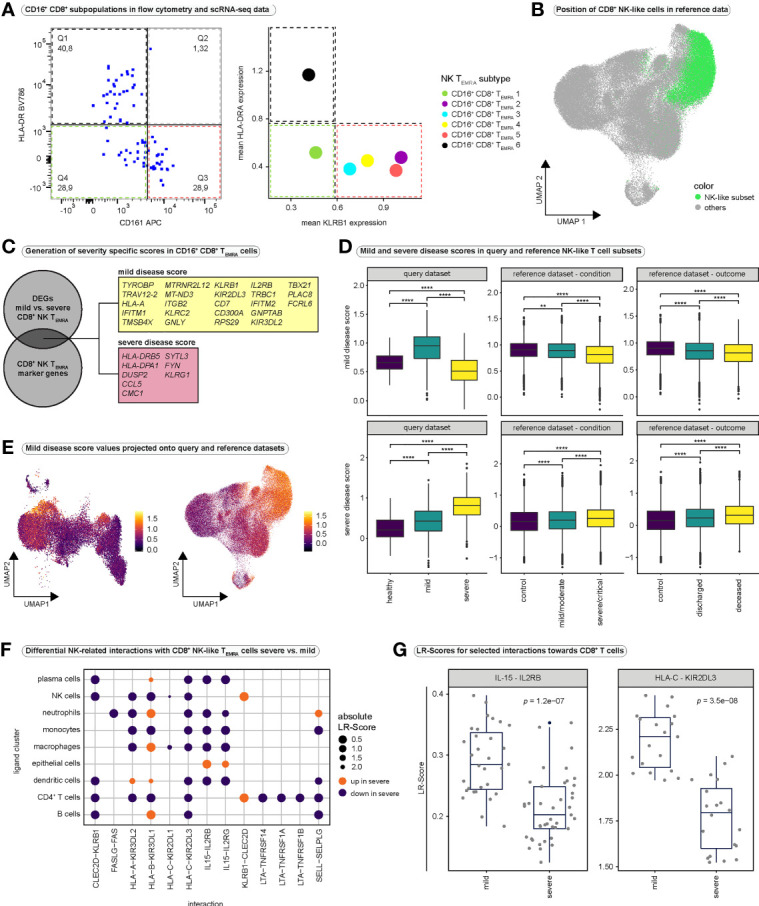
Results: We observed increased CD8+ T cell exhaustion in severe SARS-CoV-2 infection and identified a population of NK-like, terminally differentiated CD8+ effector T cells characterized by expression of FCGR3A (encoding CD16). Further characterization of NK-like CD8+ T cells revealed heterogeneity among CD16+ NK-like CD8+ T cells and profound differences in cytotoxicity, exhaustion, and NK-like differentiation between mild and severe disease conditions.
Discussion: We propose a model in which differences in the surrounding inflammatory milieu lead to crucial differences in NK-like differentiation of CD8+ effector T cells, ultimately resulting in the appearance of NK-like CD8+ T cell populations of different functionality and pathogenicity. Our in-depth characterization of the CD8+ T cell-mediated response to SARS-CoV-2 infection provides a basis for further investigation of the importance of NK-like CD8+ T cells in COVID-19 severity.
Keywords: CD16; CD8+ T cells; FCGR3A; NK-like T cell; SARS-CoV-2; immunoprofiling; scRNA-seq; scTCR-seq.
Copyright © 2022 Schreibing, Hannani, Kim, Nagai, Ticconi, Fewings, Bleckwehl, Begemann, Torow, Kuppe, Kurth, Kranz, Frank, Anslinger, Ziegler, Kraus, Enczmann, Balz, Windhofer, Balfanz, Kurts, Marx, Marx, Dreher, Schneider, Saez-Rodriguez, Costa, Hayat and Kramann.
Conflict of interest statement
Author JS-R declares funding from GSK and Sanofi and consultant fees from Travere Therapeutics, Owkin and Astex Pharmaceuticals unrelated to this study. Author SH has funding from Novo Nordisk. Author RK has grants from Travere Therapeutics, Galapagos, Chugai, and Novo Nordisk and is a consultant or received honoraria from Bayer, Pfizer, Novo Nordisk, Lilly-Pharma and Gruenenthal. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
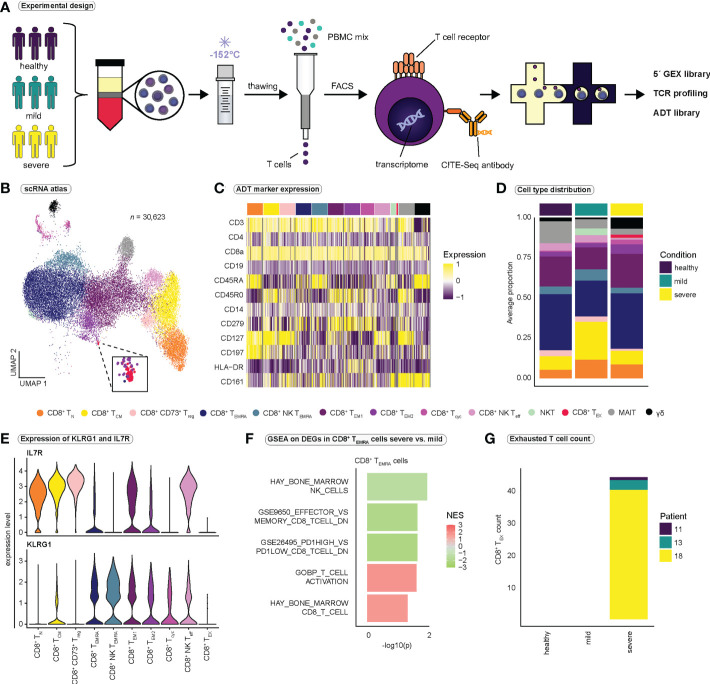
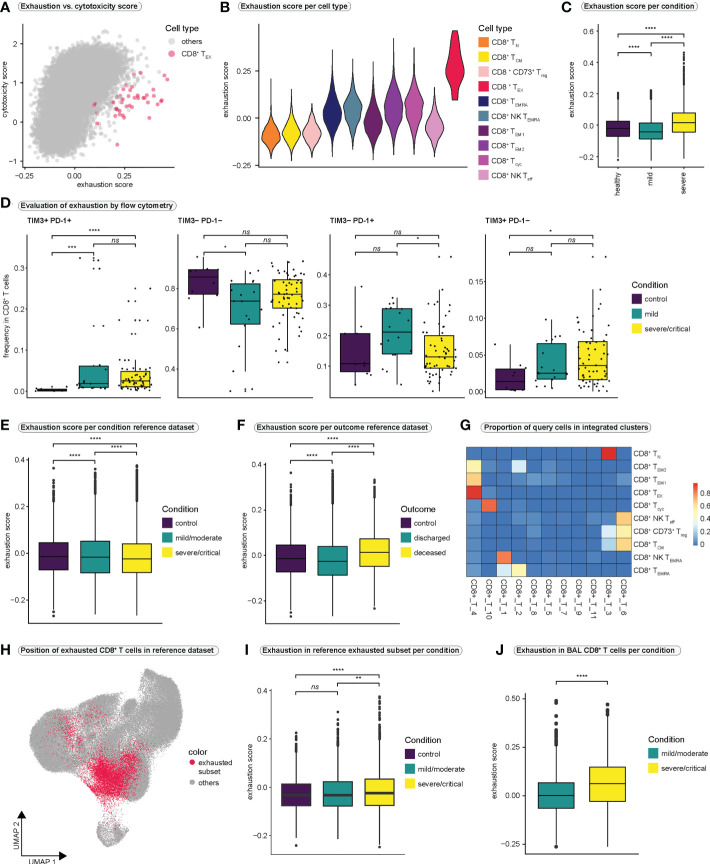
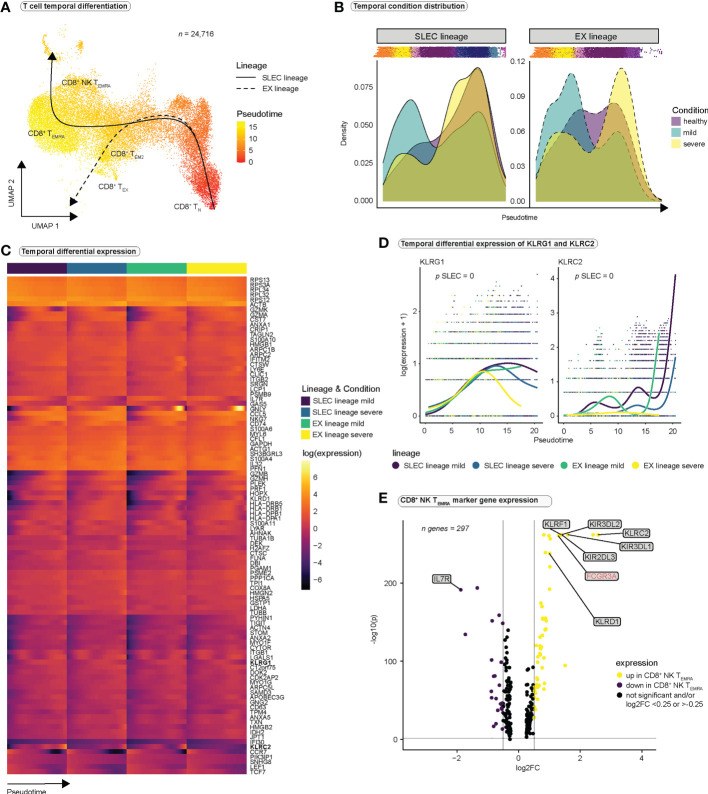
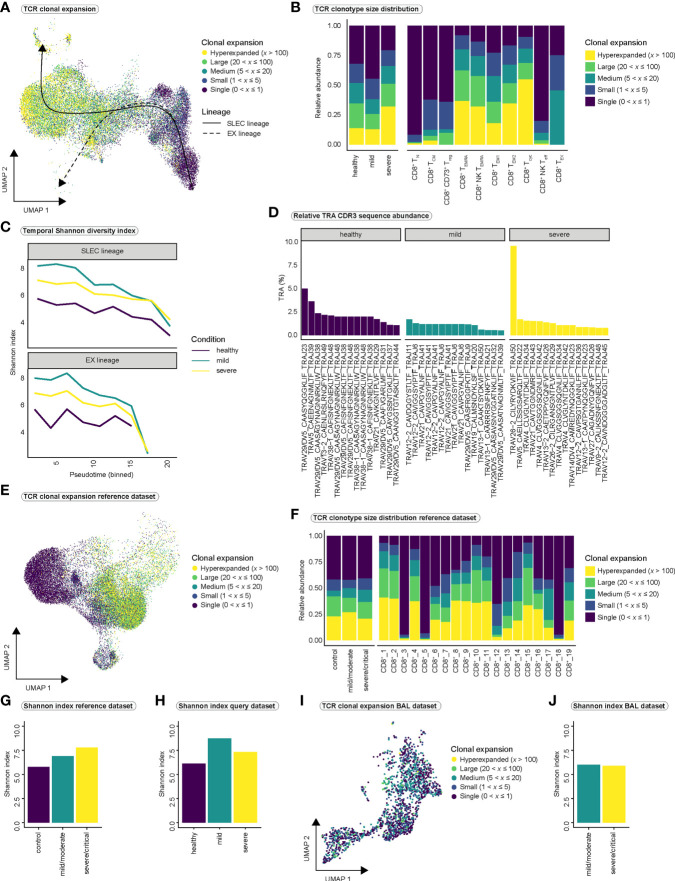
References
-
- WHO . Coronavirus (COVID-19) dashboard. Available at: https://covid19.who.int/ (Accessed August 23, 2022).
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous