

A proteome-wide atlas of drug mechanism of action
- PMID: 36593396
- PMCID: PMC11069389
- DOI: 10.1038/s41587-022-01539-0
A proteome-wide atlas of drug mechanism of action
Abstract
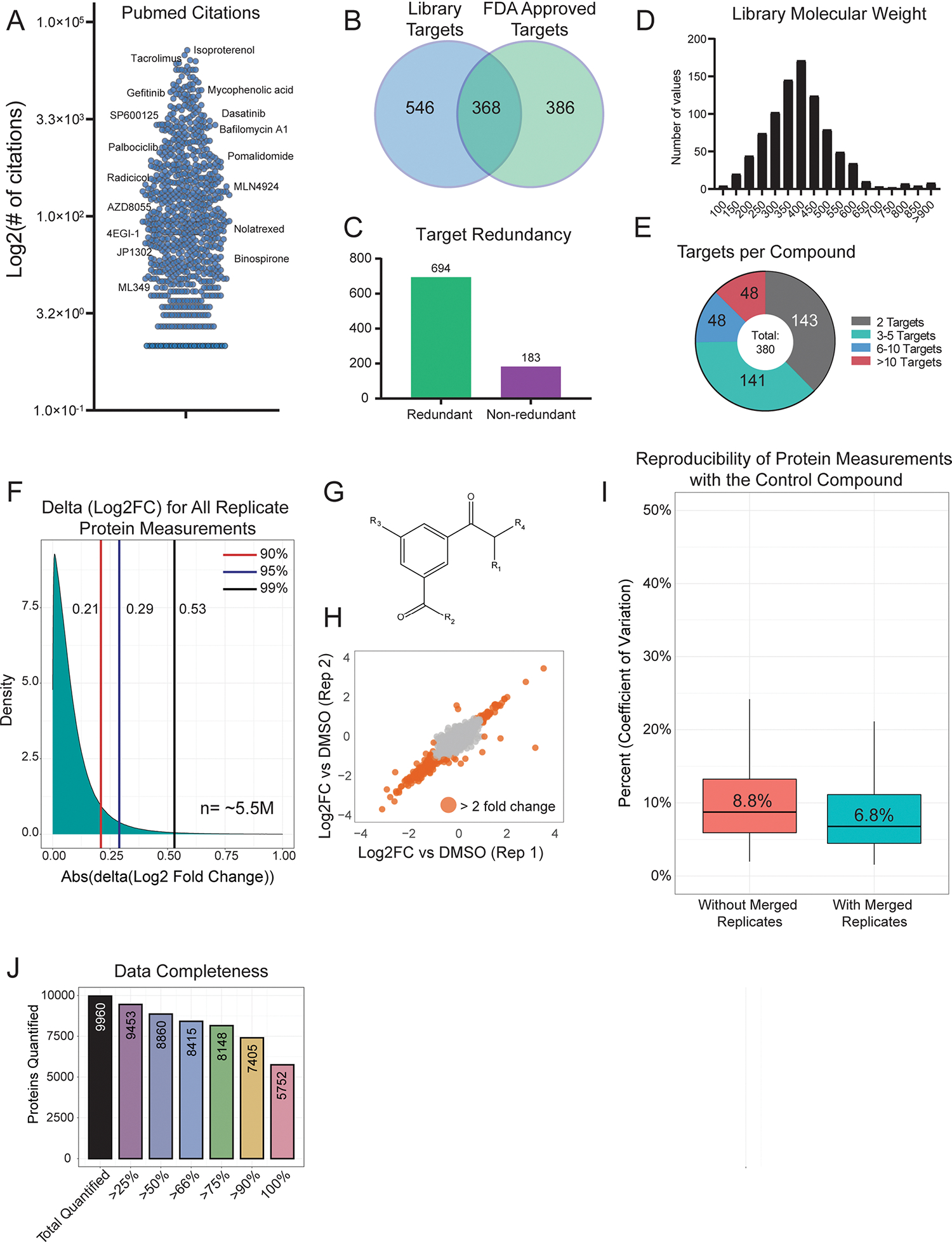
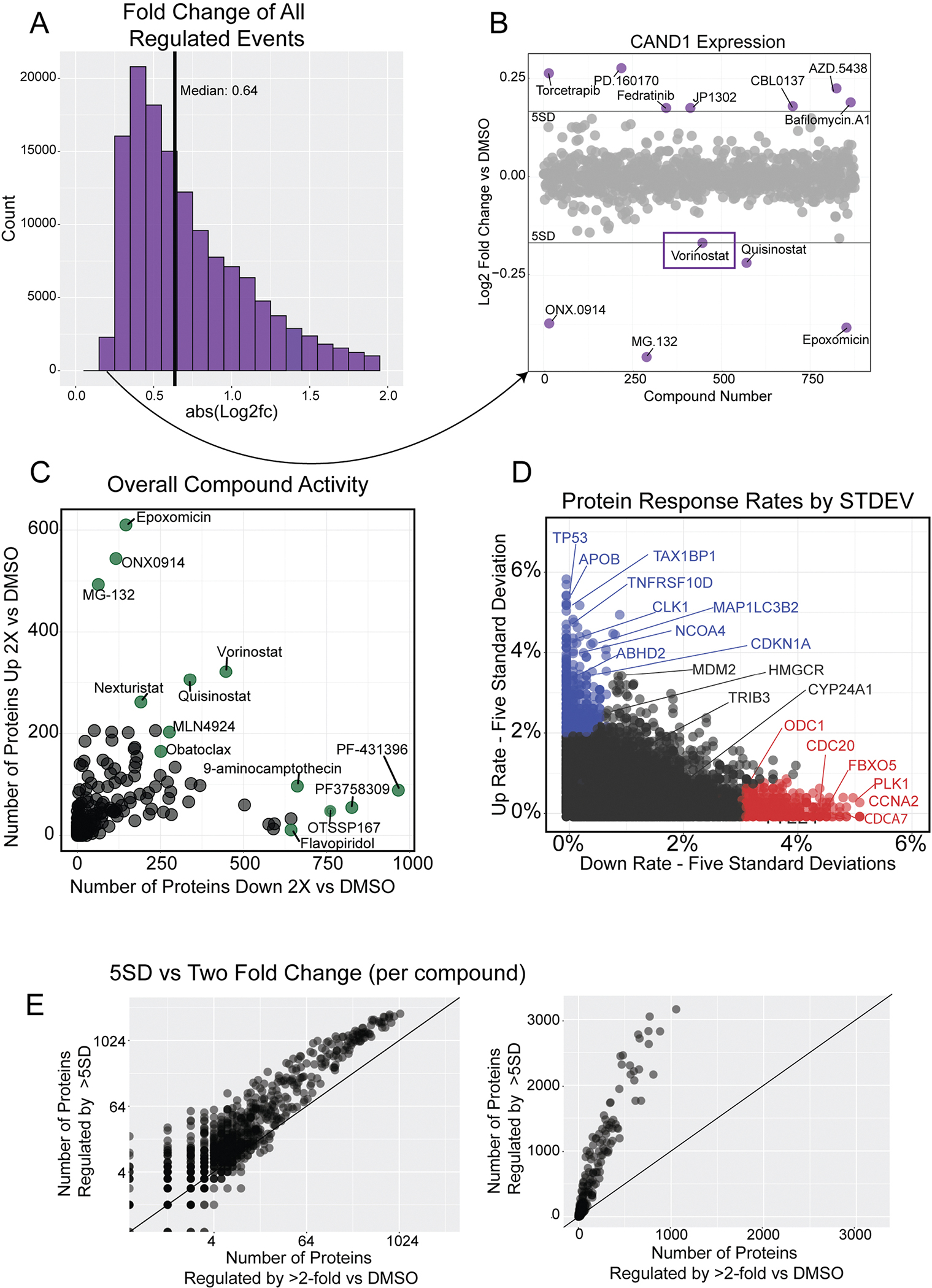
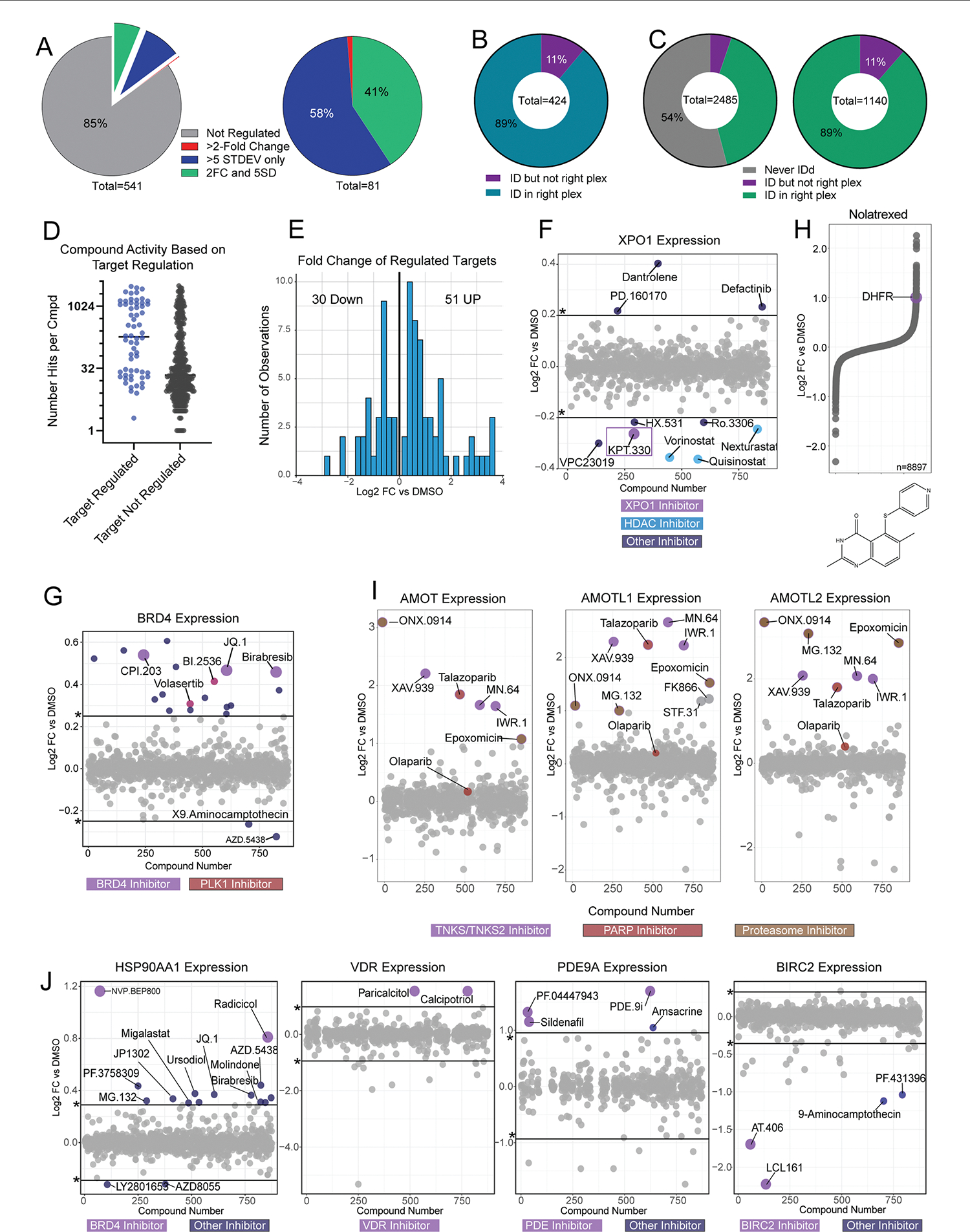
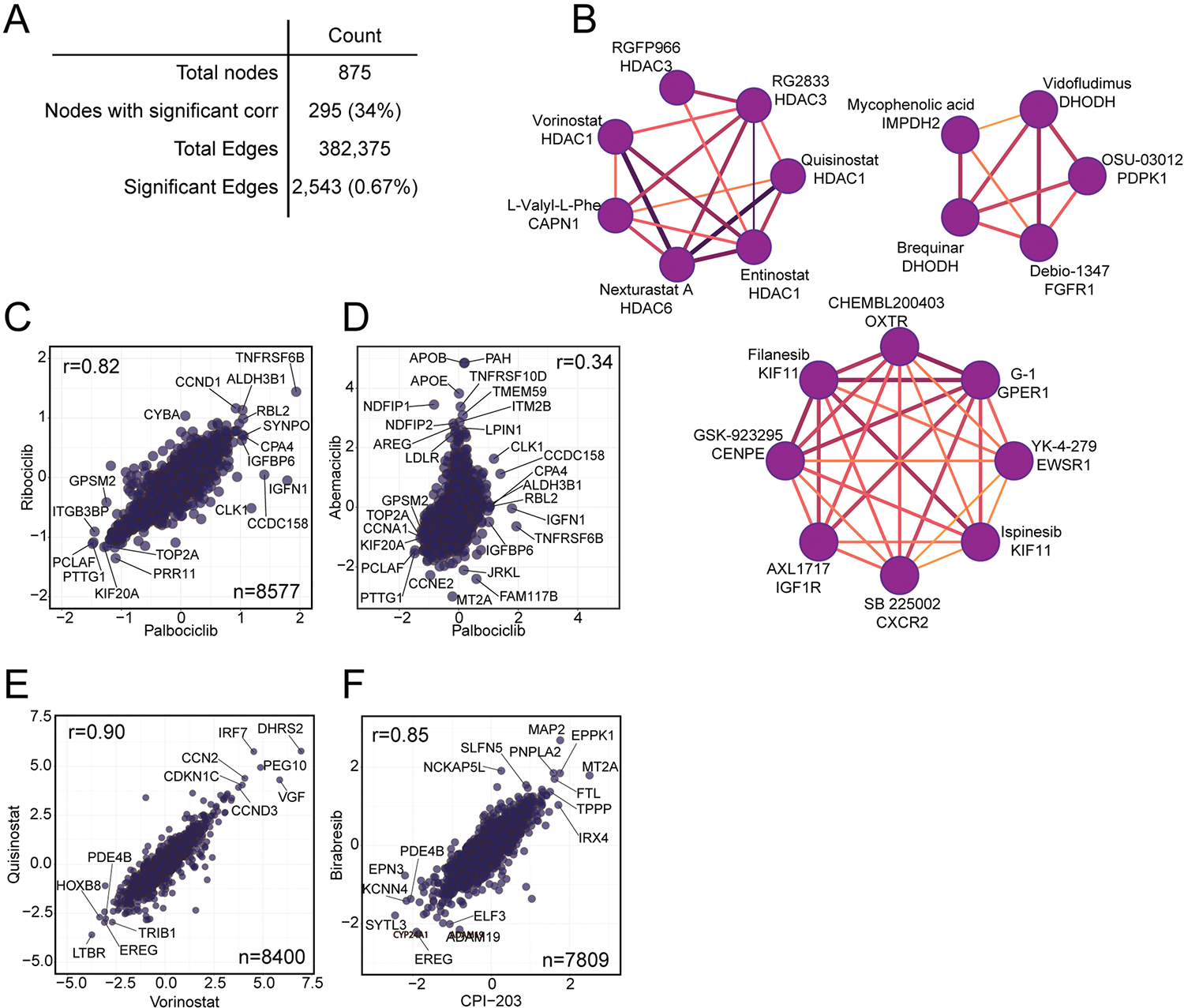
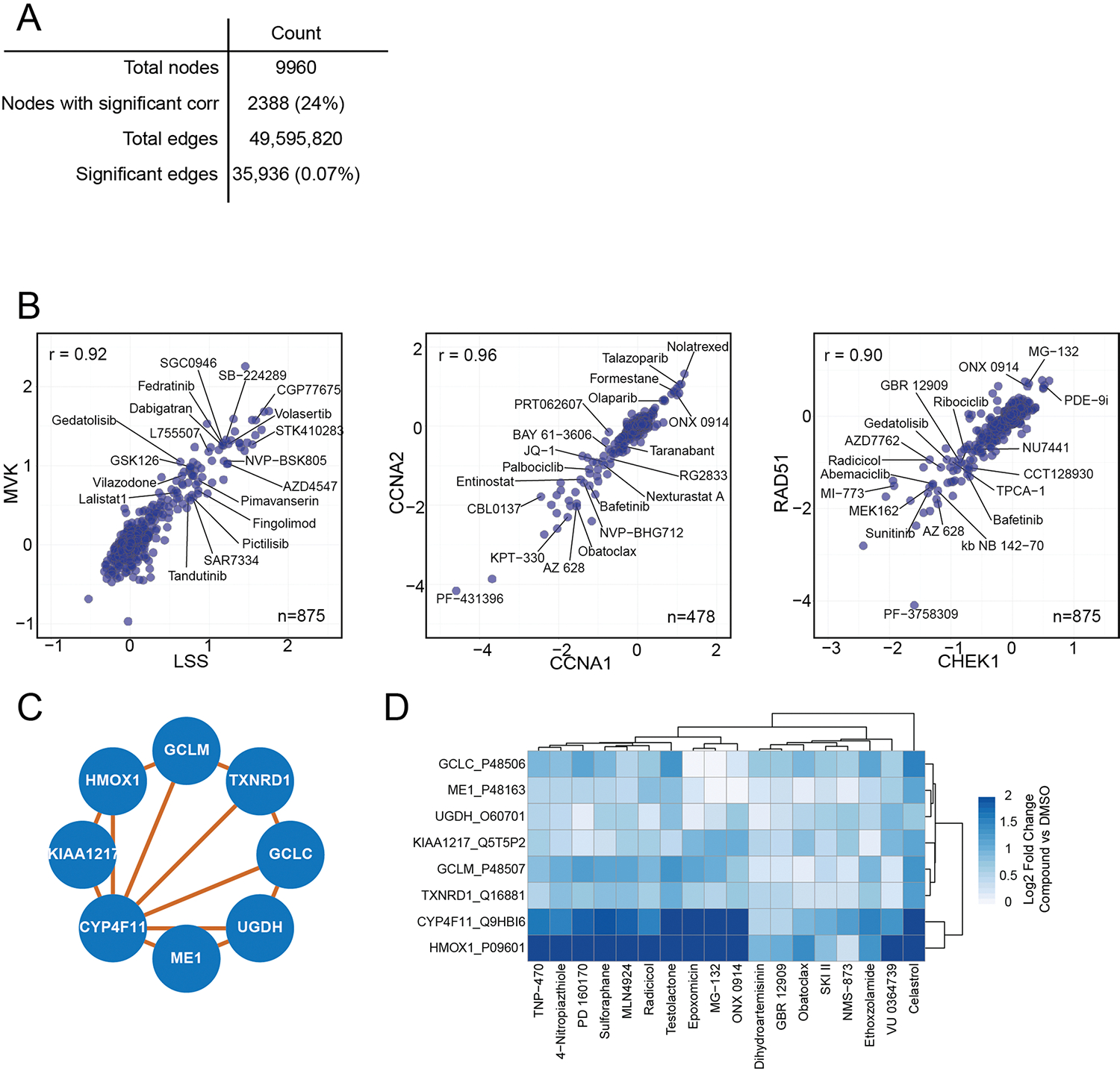
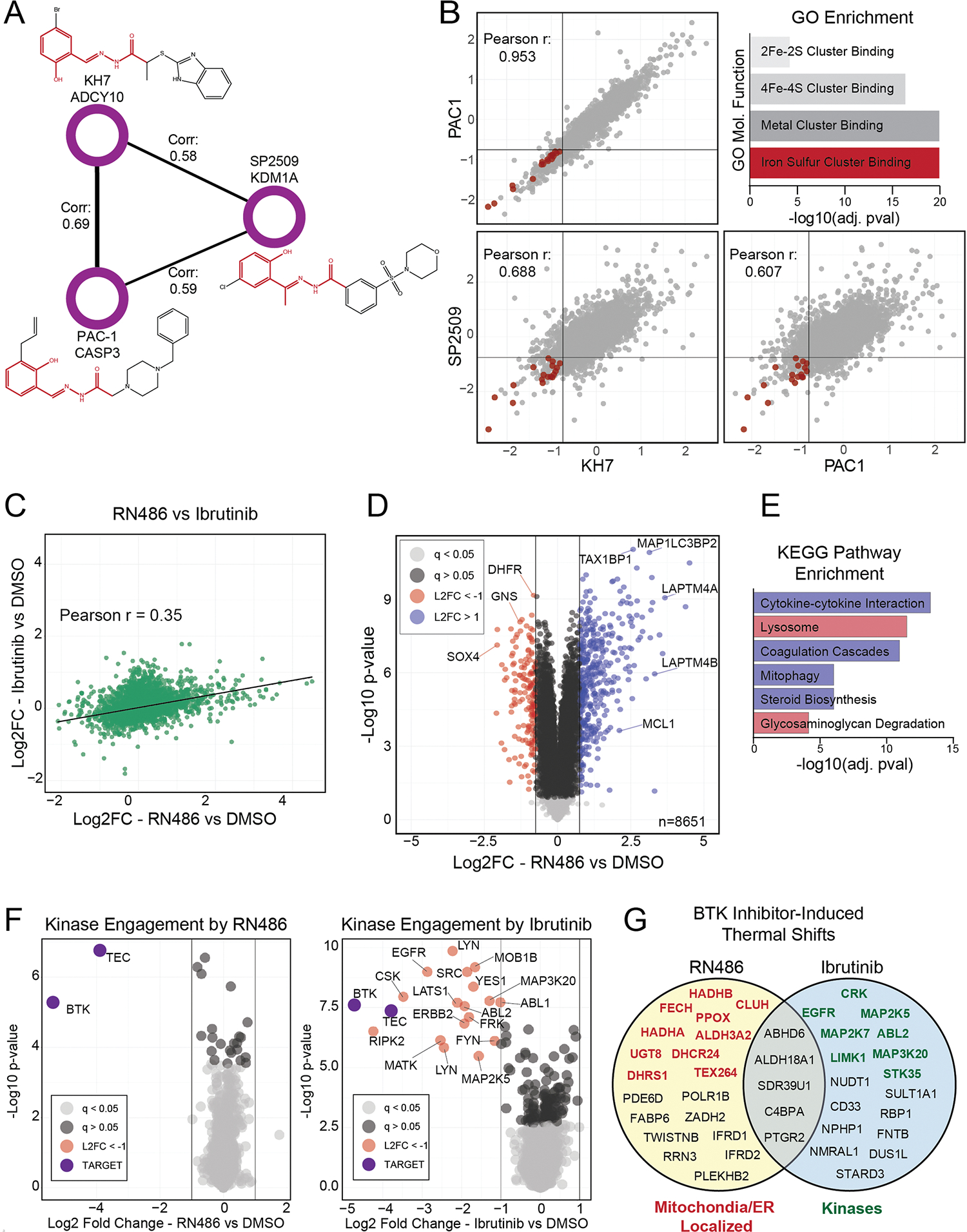
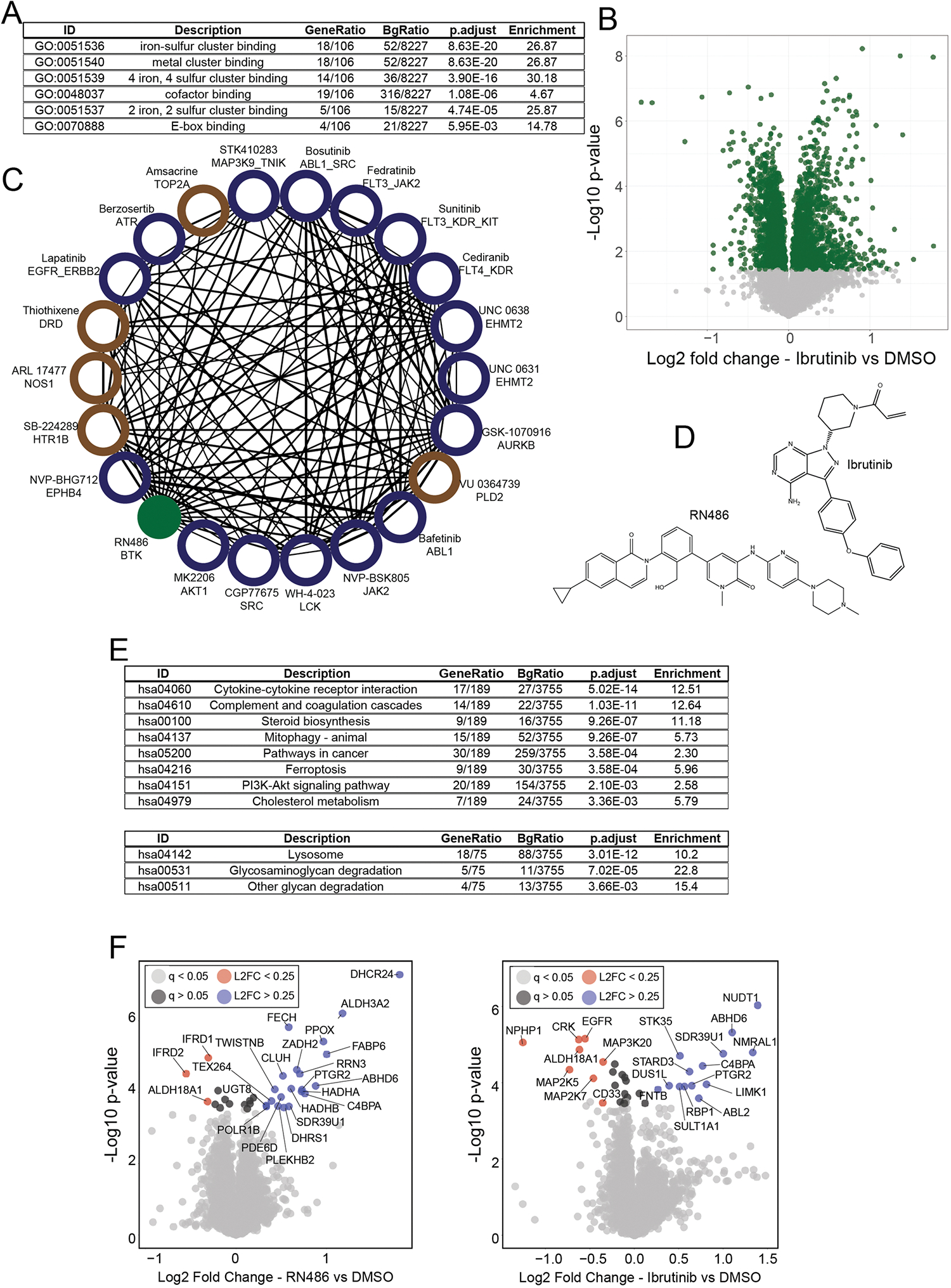
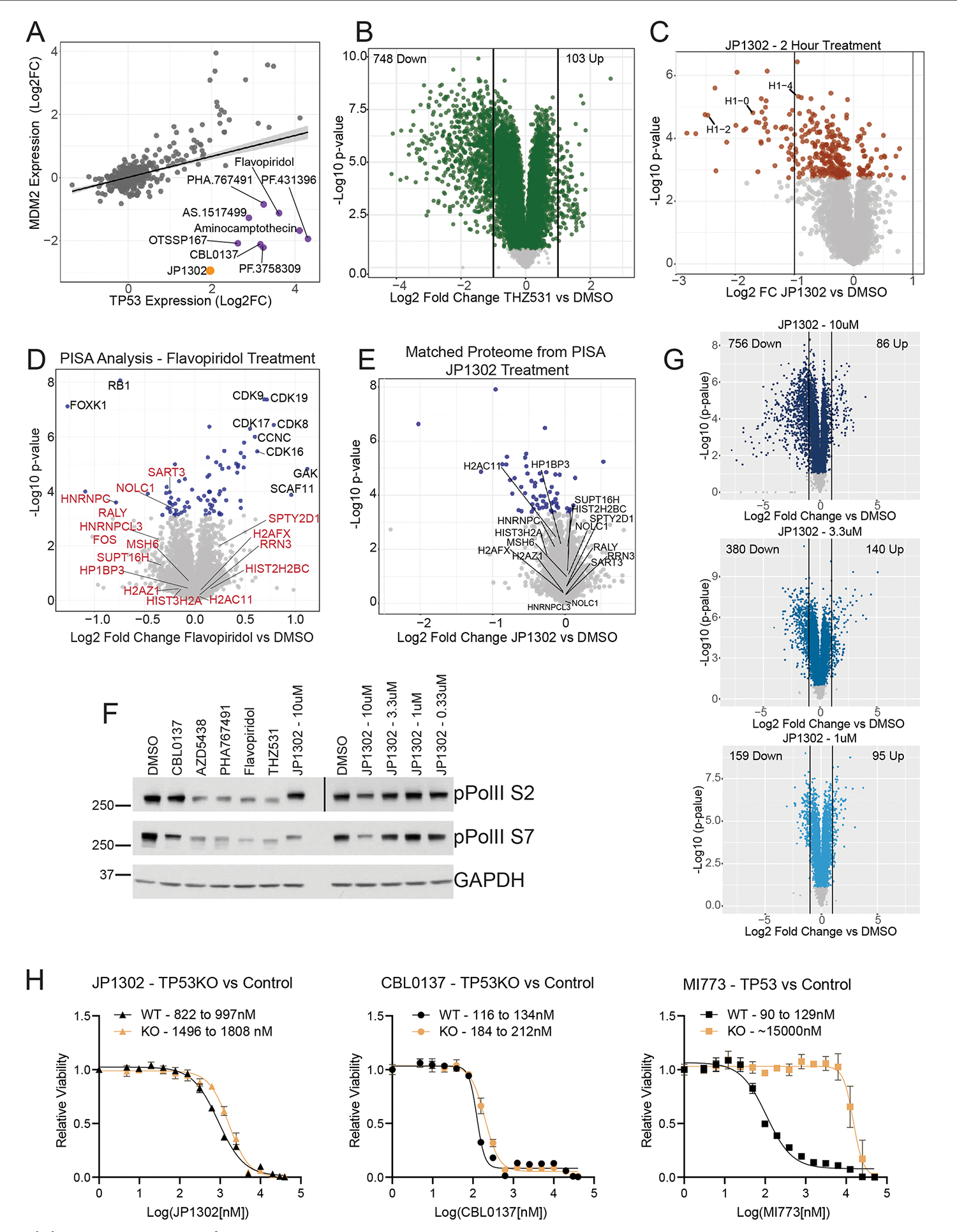
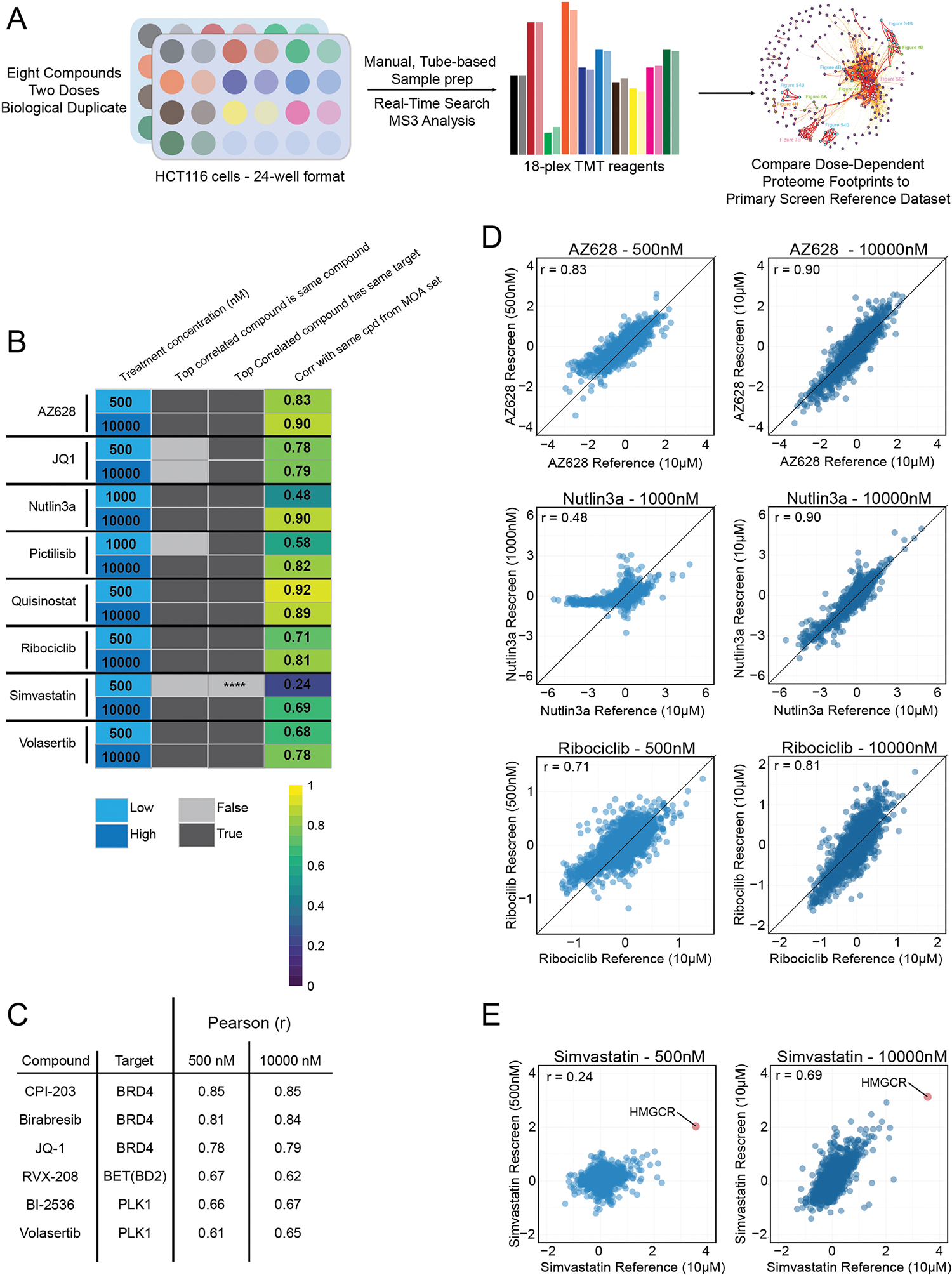
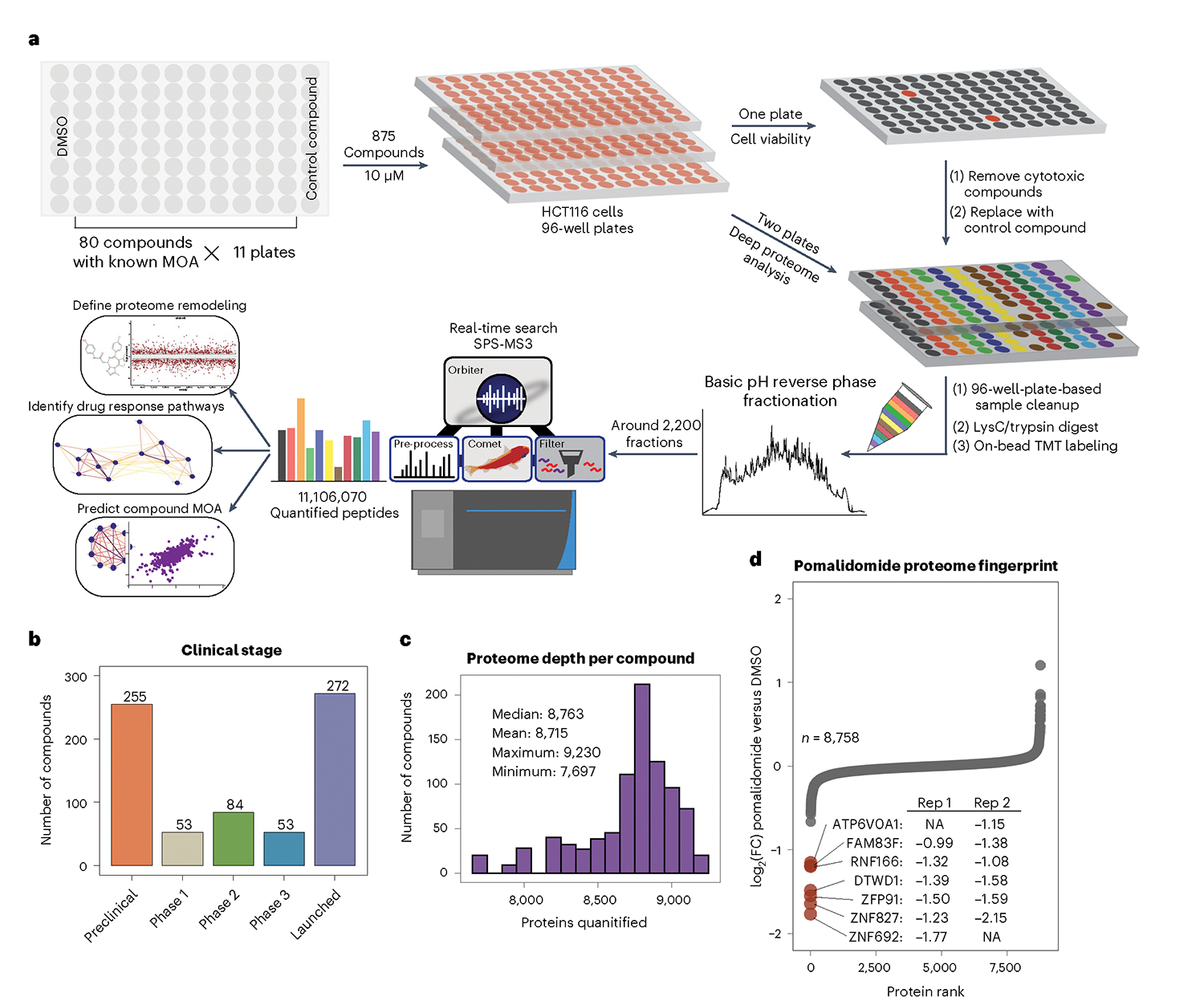
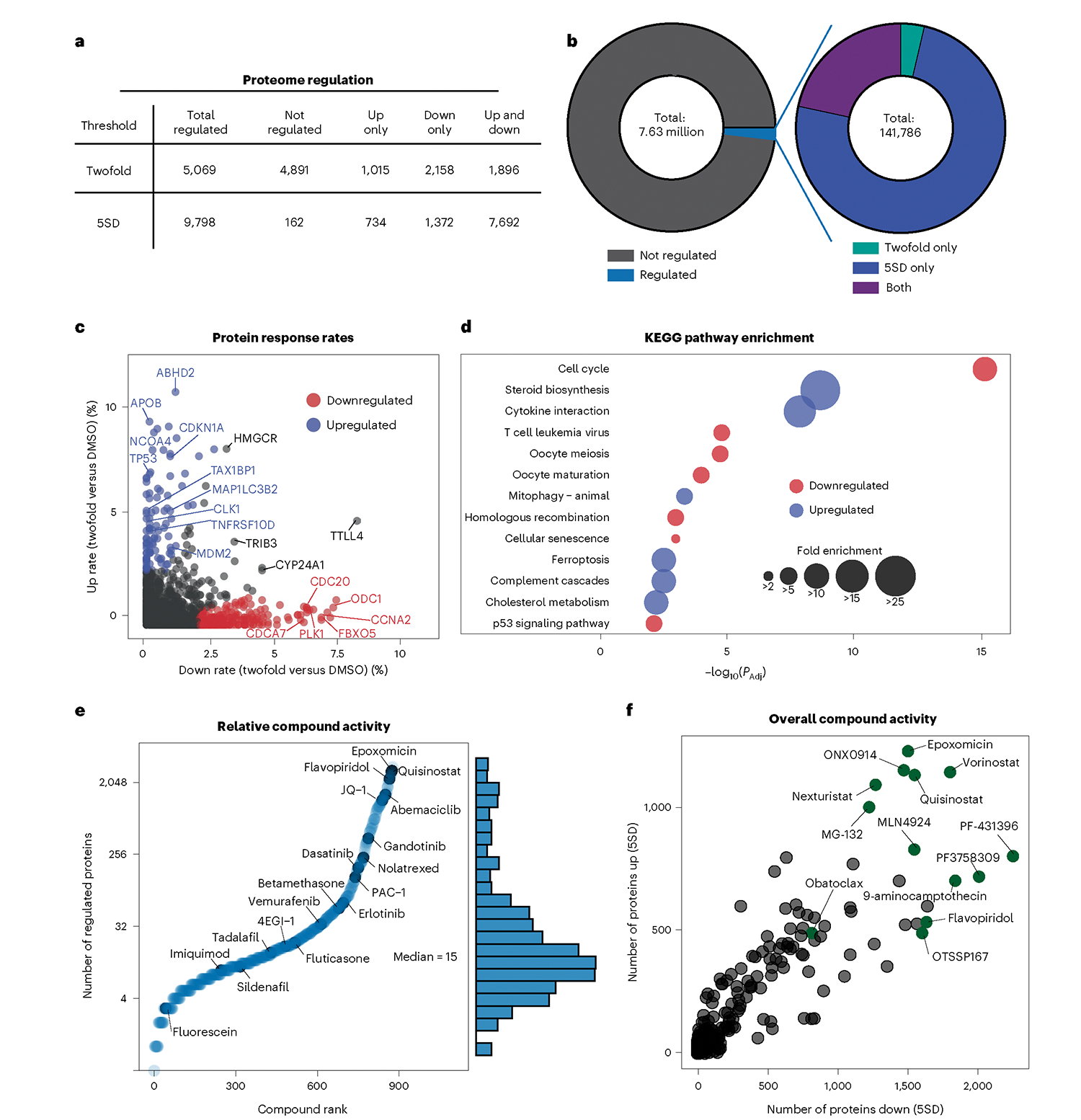
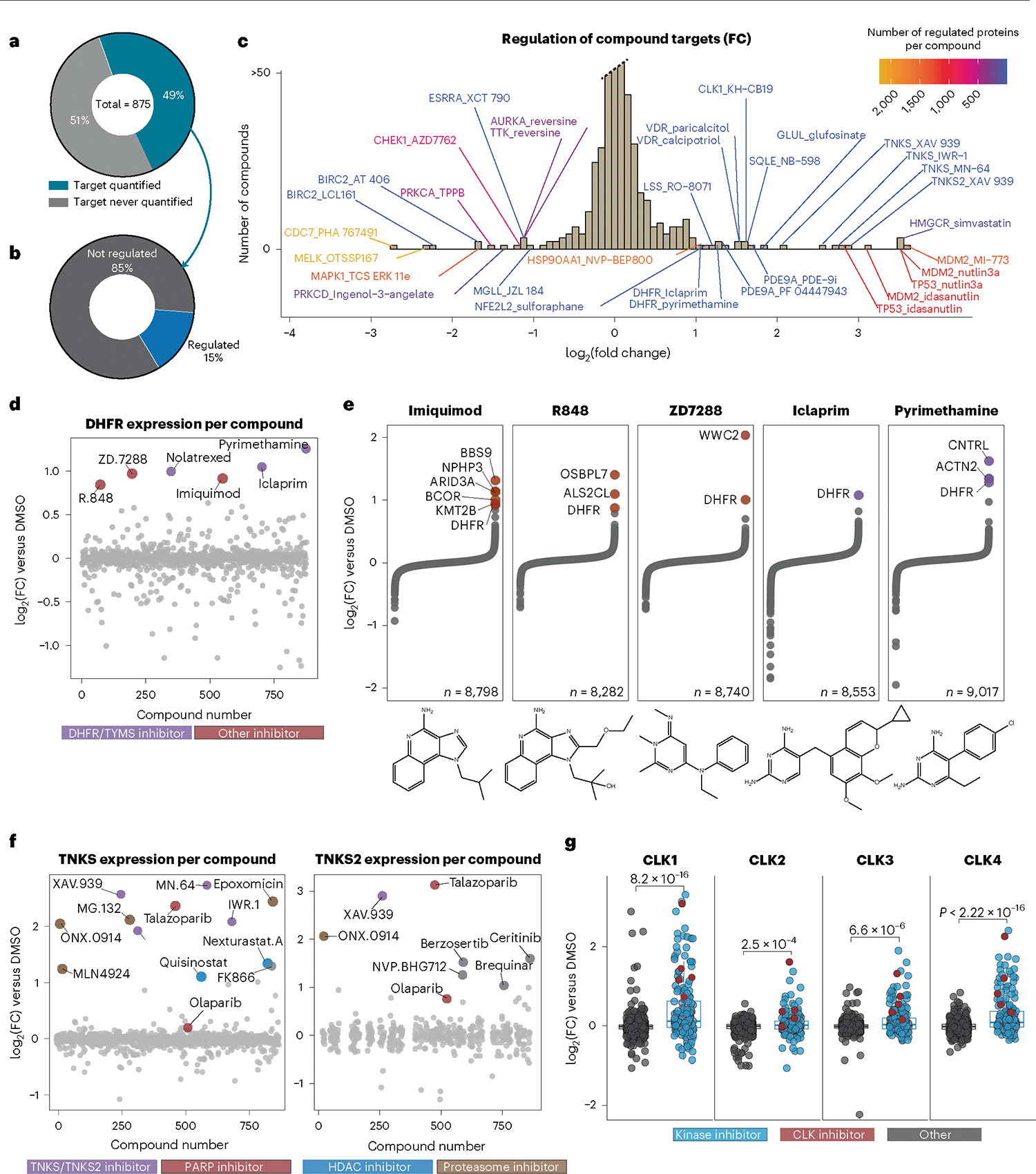
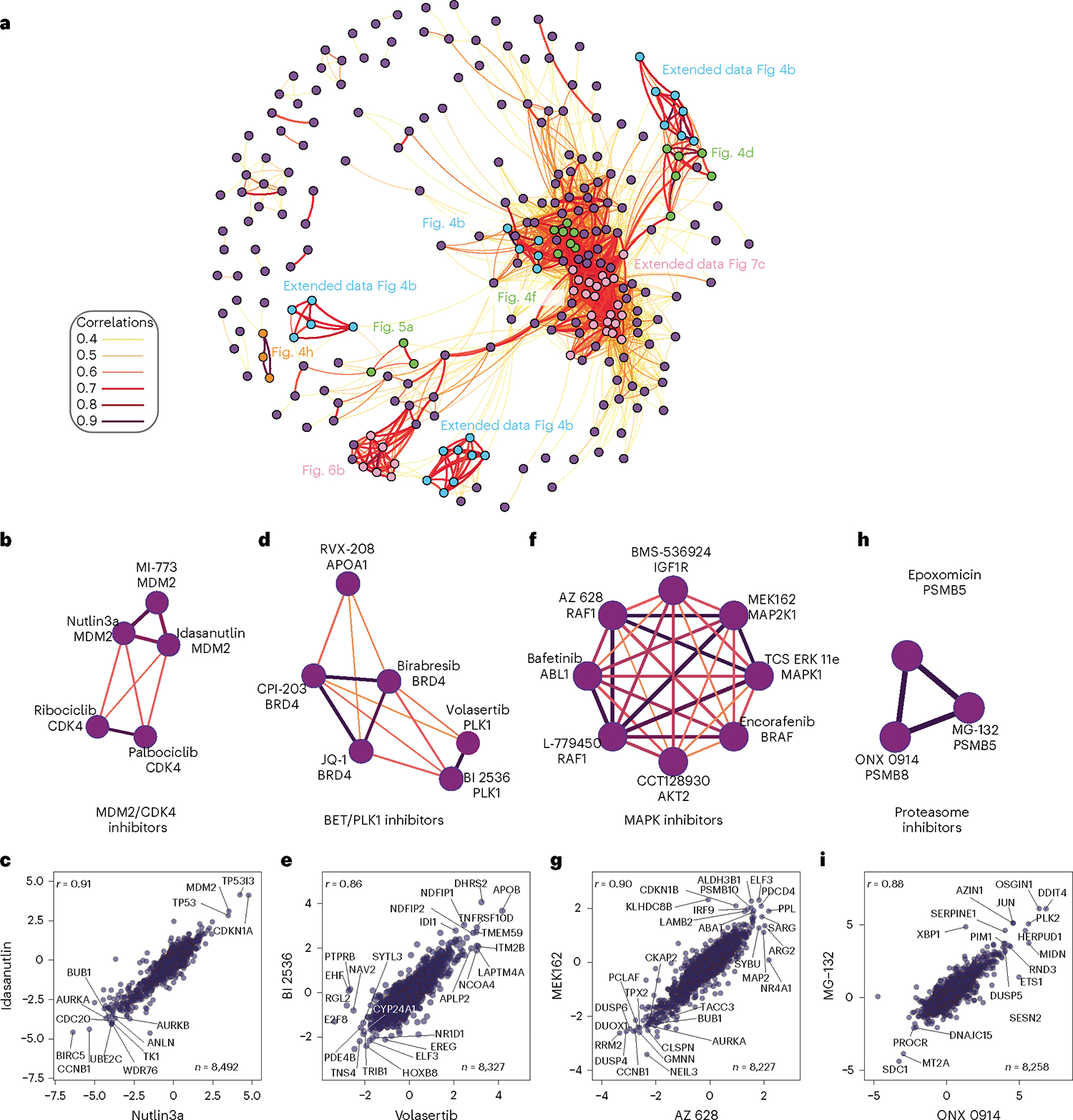
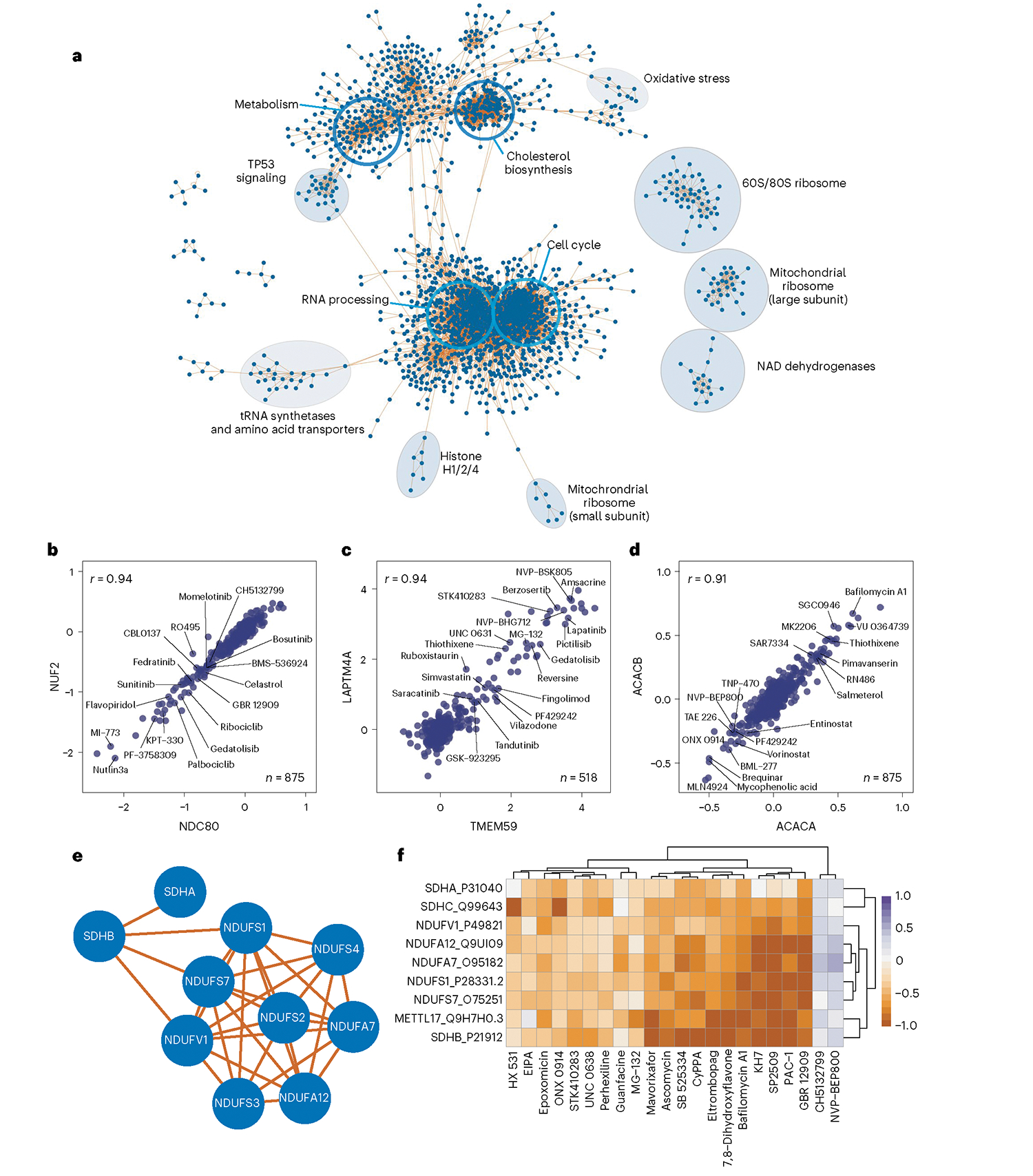
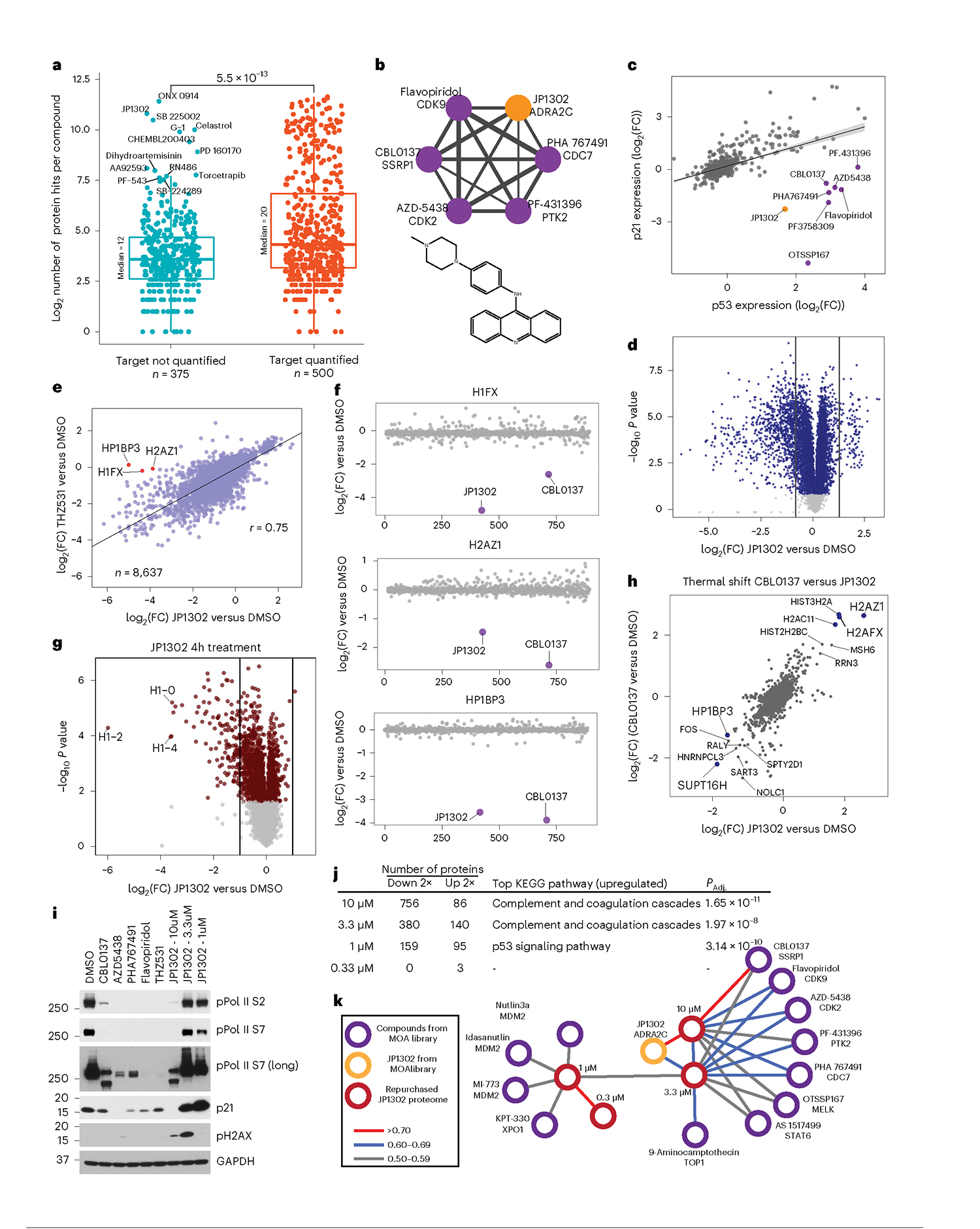
Defining the cellular response to pharmacological agents is critical for understanding the mechanism of action of small molecule perturbagens. Here, we developed a 96-well-plate-based high-throughput screening infrastructure for quantitative proteomics and profiled 875 compounds in a human cancer cell line with near-comprehensive proteome coverage. Examining the 24-h proteome changes revealed ligand-induced changes in protein expression and uncovered rules by which compounds regulate their protein targets while identifying putative dihydrofolate reductase and tankyrase inhibitors. We used protein-protein and compound-compound correlation networks to uncover mechanisms of action for several compounds, including the adrenergic receptor antagonist JP1302, which we show disrupts the FACT complex and degrades histone H1. By profiling many compounds with overlapping targets covering a broad chemical space, we linked compound structure to mechanisms of action and highlighted off-target polypharmacology for molecules within the library.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
S.P.G. is an advisory board member for Cell Signaling Technology, ThermoFisher Scientific, Cedilla Therapeutics, Casma Therapeutics and Frontier Medicines. The remaining authors declare no competing interests.
Figures
Comment in
-
Proteomics reveals small molecules' secrets.Nat Rev Drug Discov. 2023 Mar;22(3):181. doi: 10.1038/d41573-023-00019-7. Nat Rev Drug Discov. 2023. PMID: 36747052 No abstract available.
References
-
- Liu Y, Beyer A & Aebersold R On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
