

High-throughput, targeted MHC class I immunopeptidomics using a functional genetics screening platform
- PMID: 36593401
- PMCID: PMC10314971
- DOI: 10.1038/s41587-022-01566-x
High-throughput, targeted MHC class I immunopeptidomics using a functional genetics screening platform
Abstract
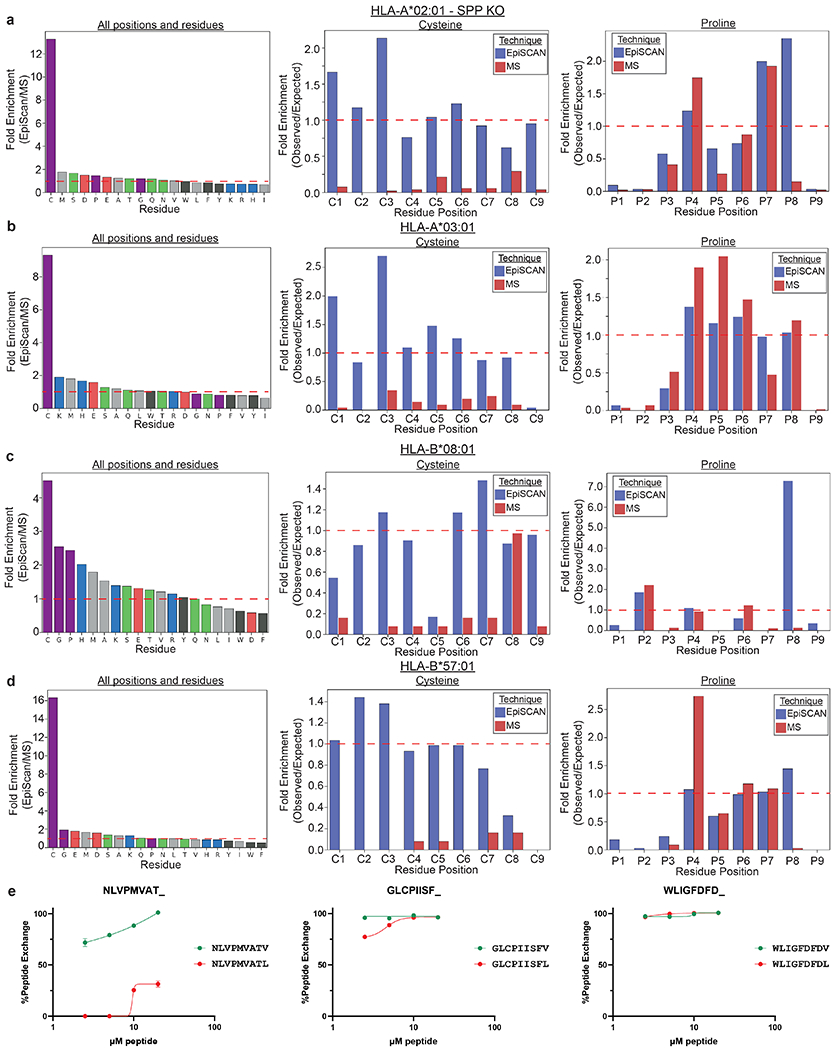
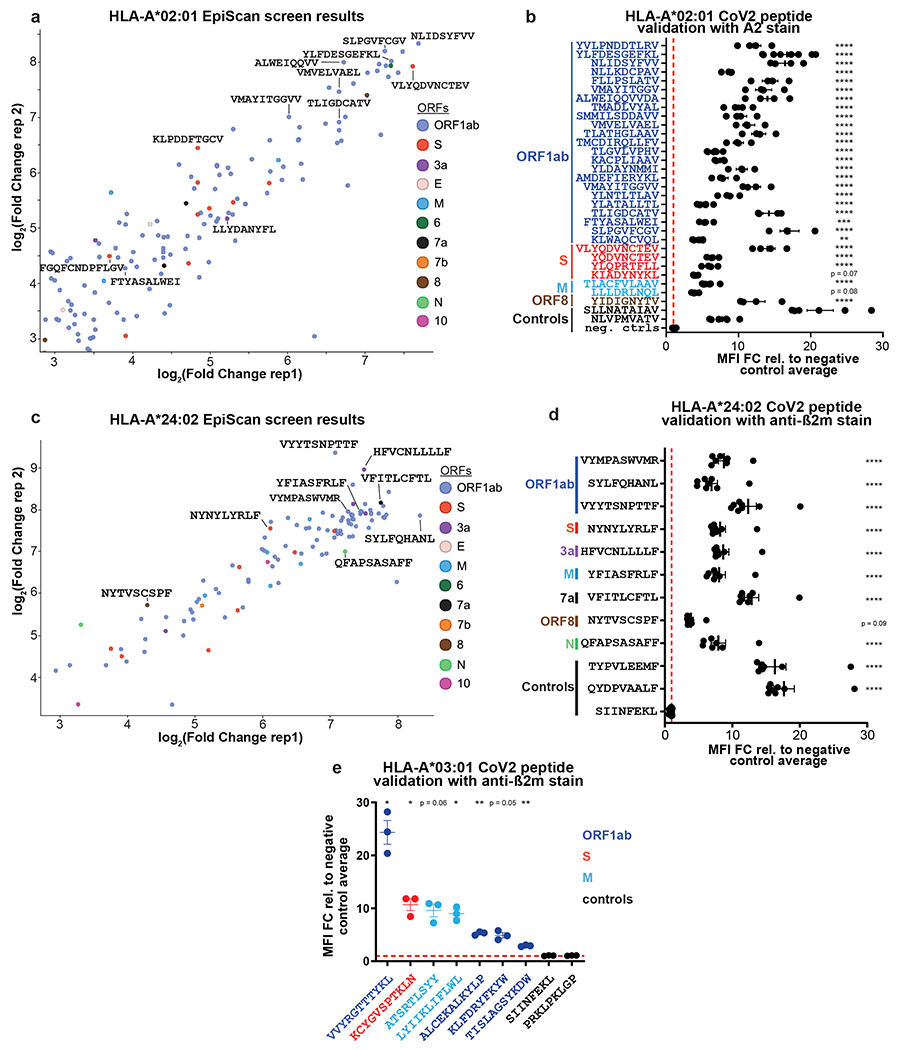
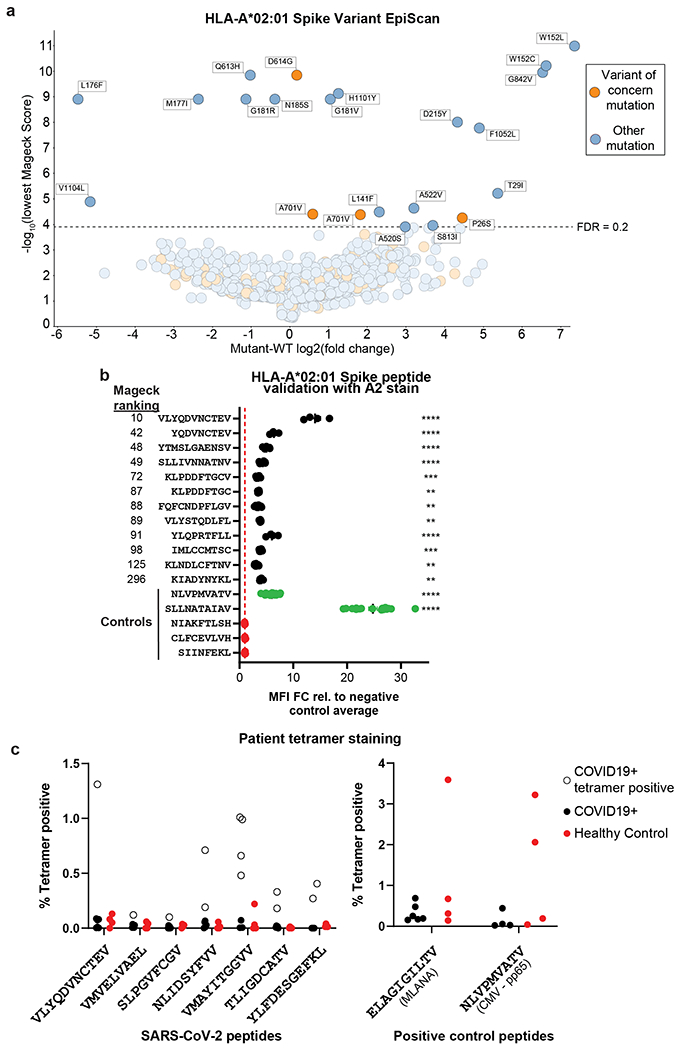
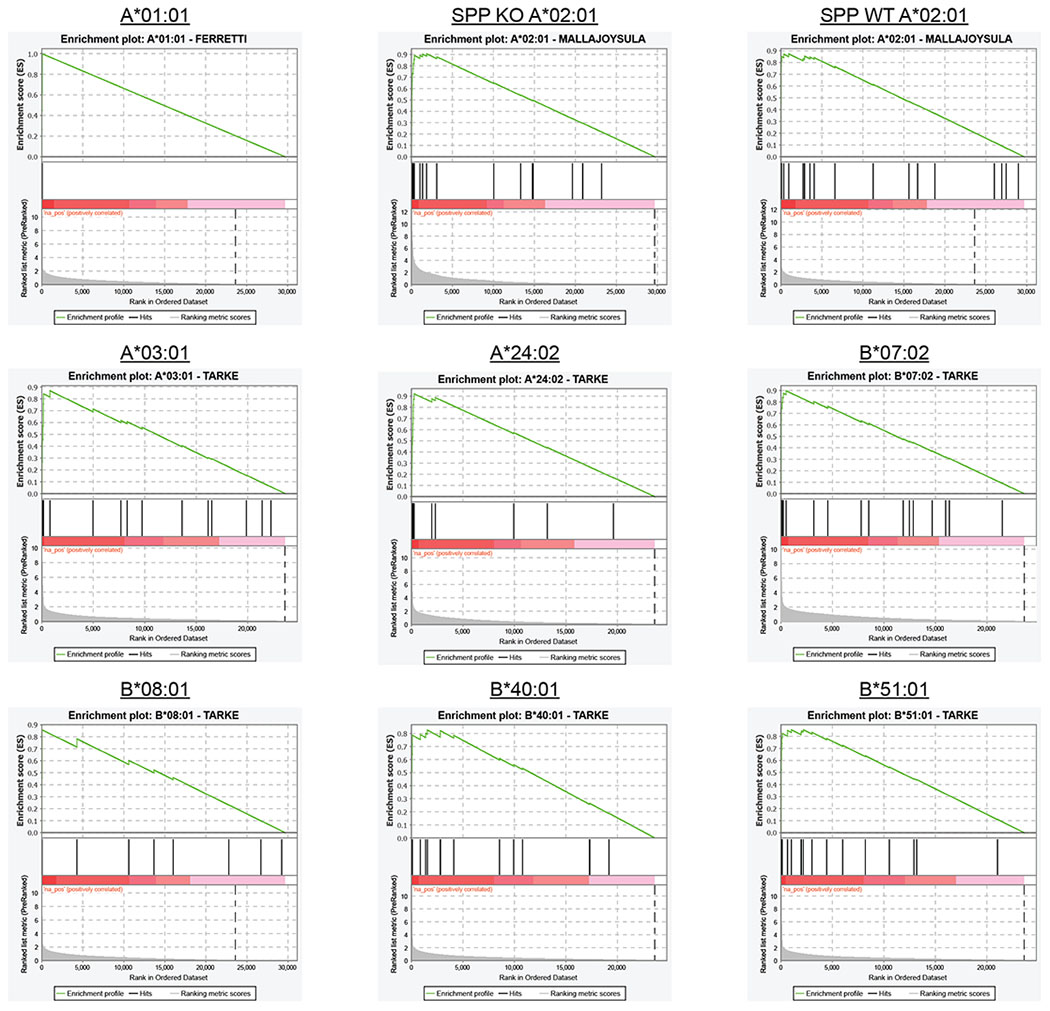
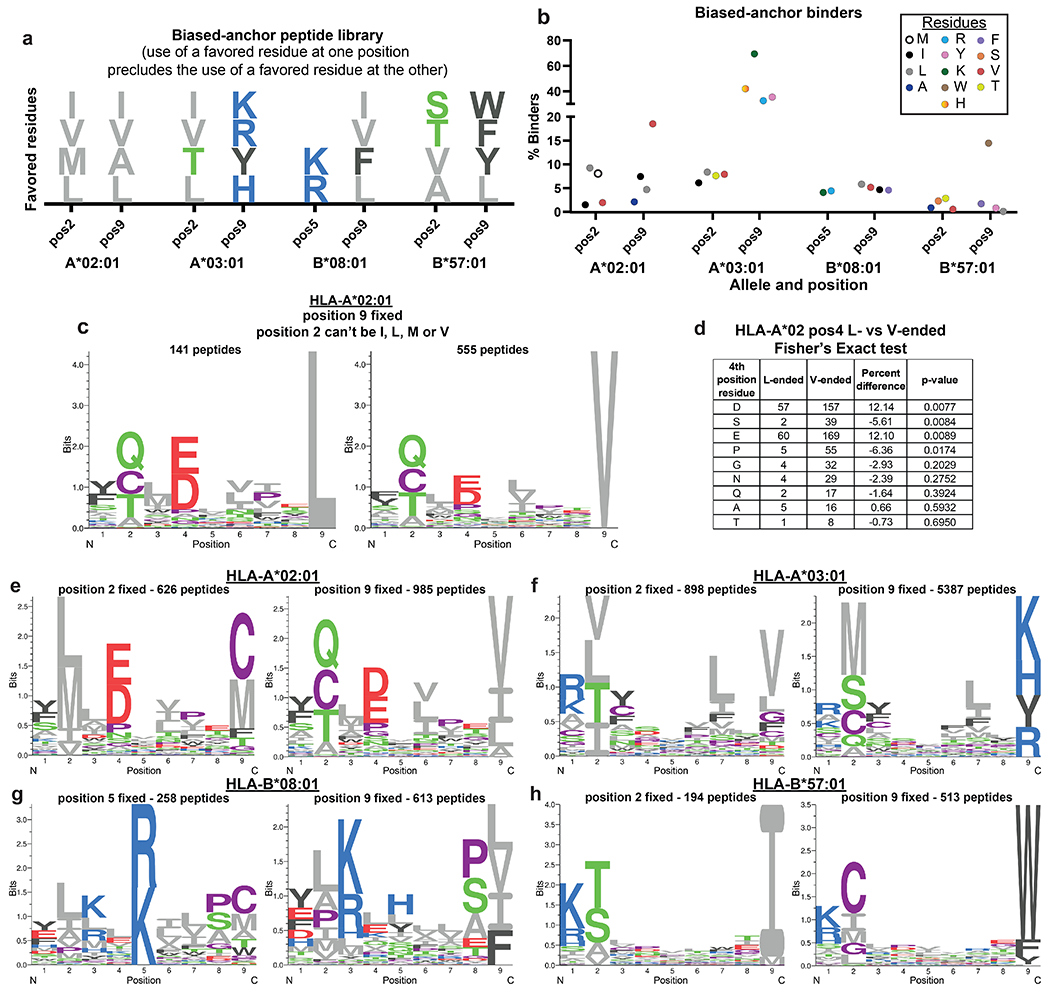
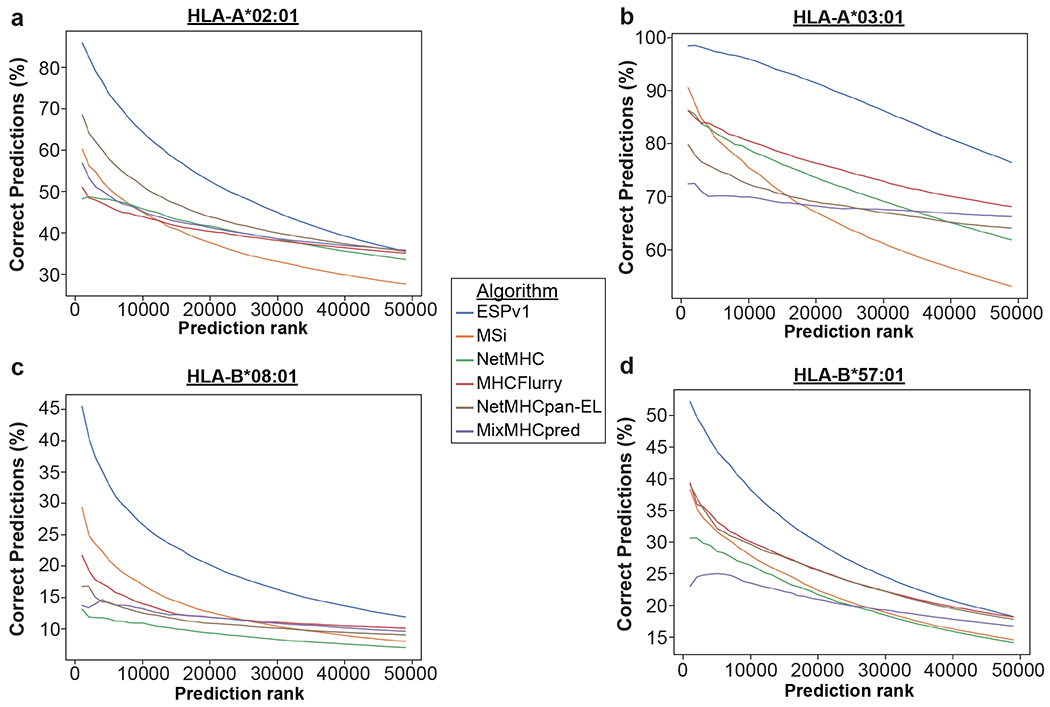
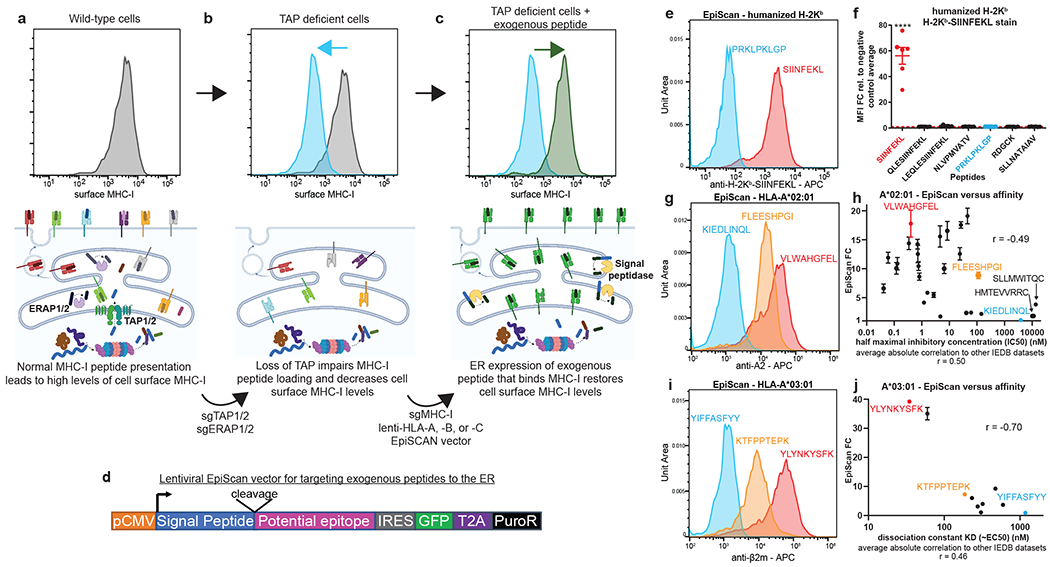
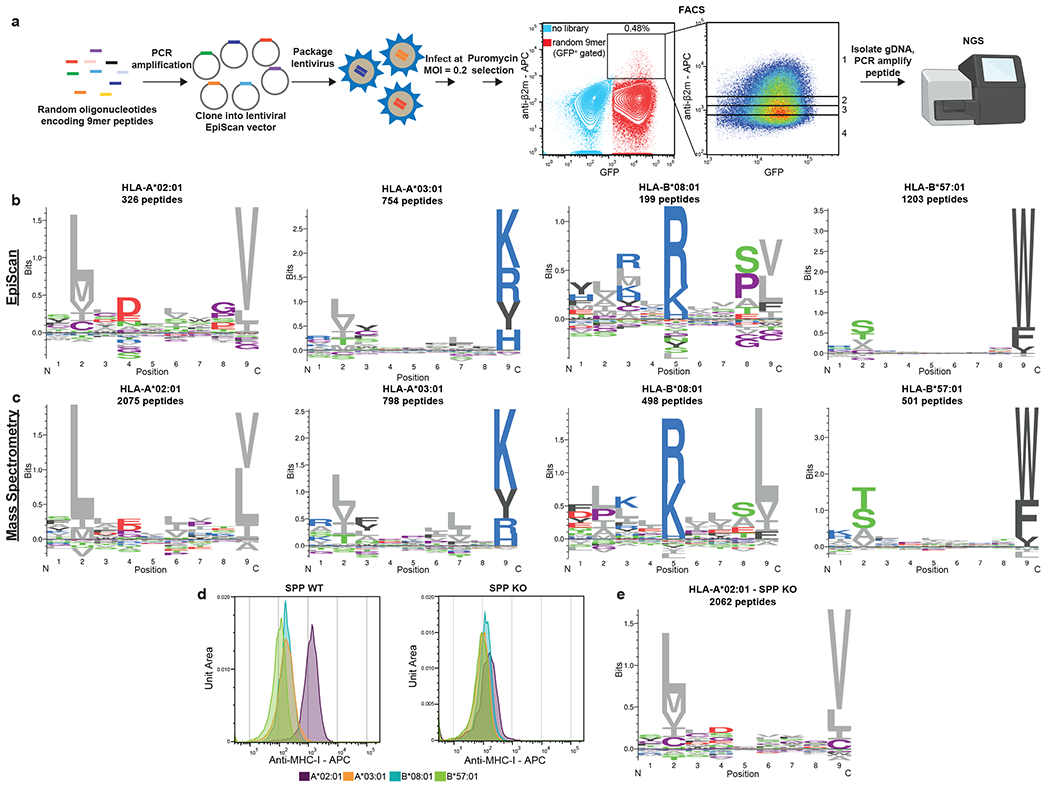
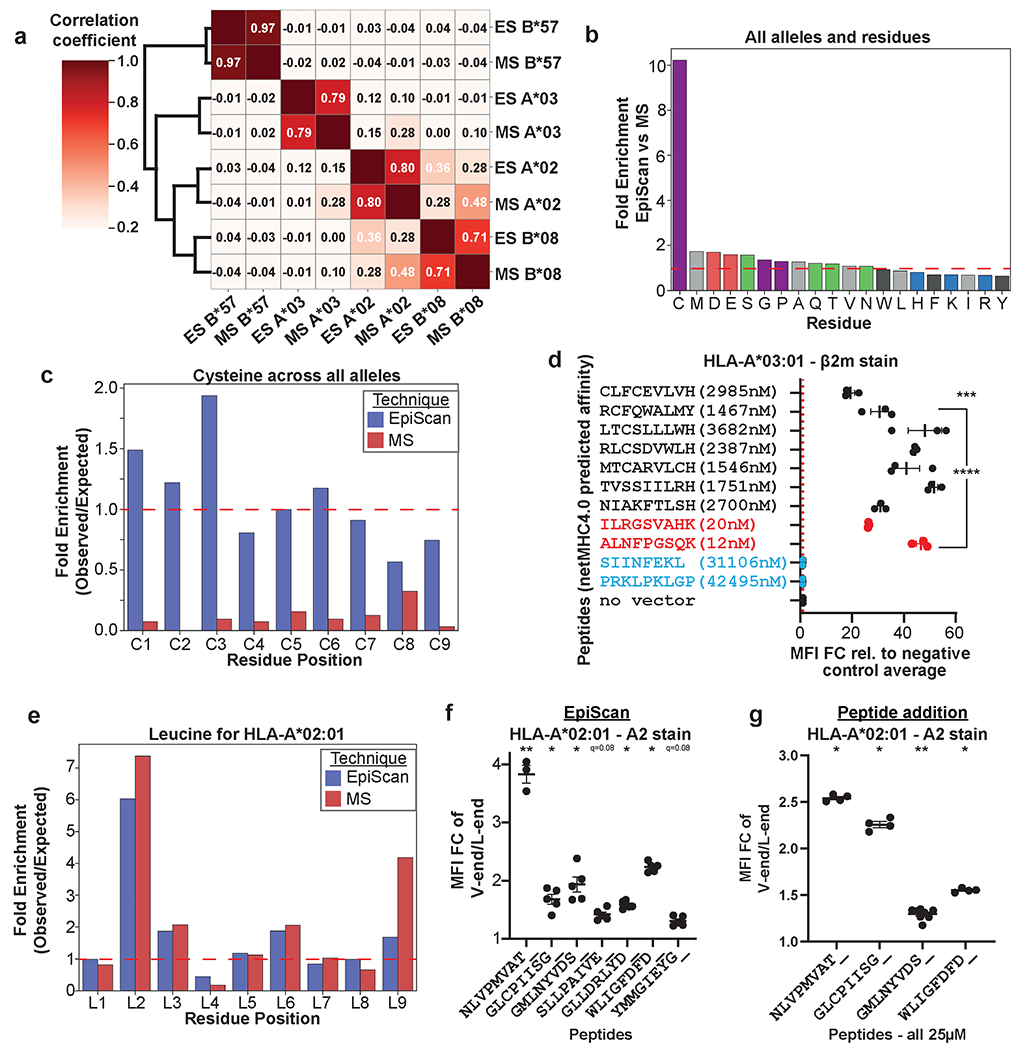
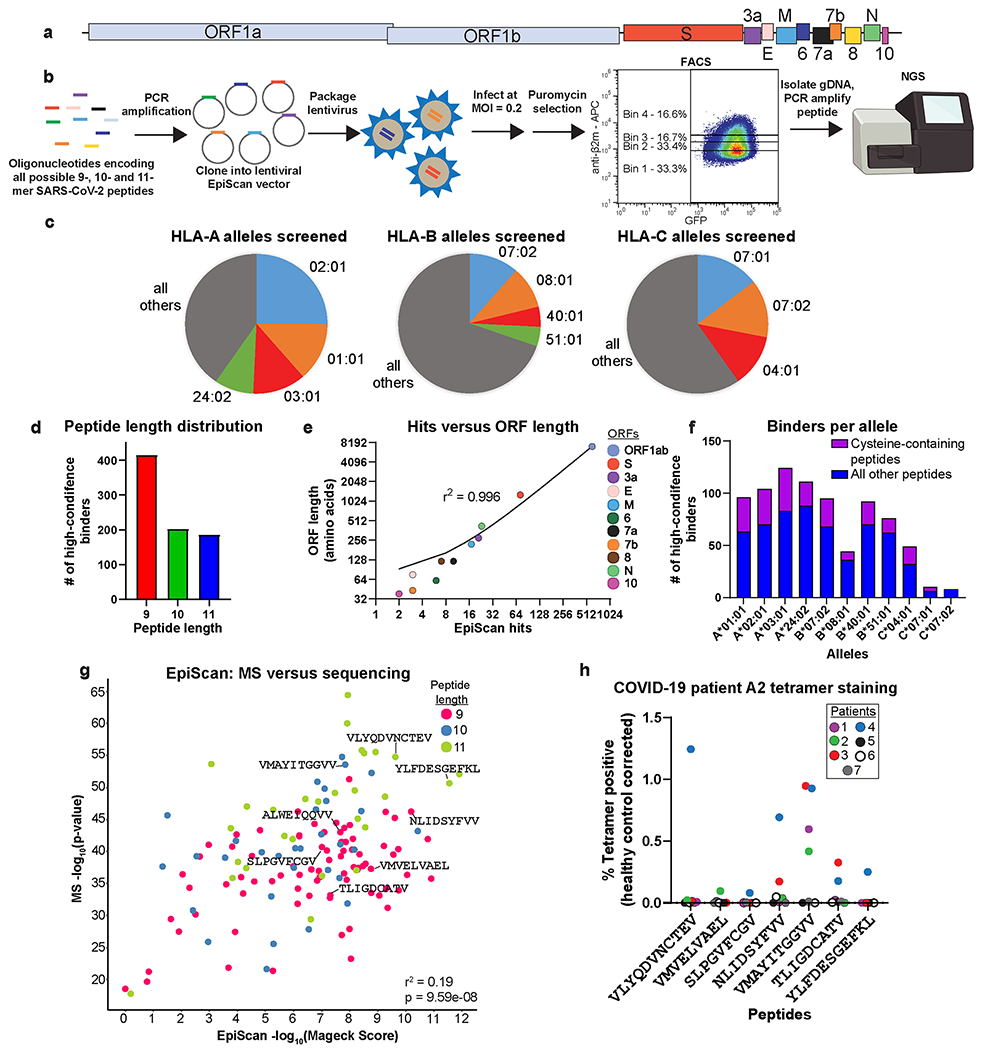
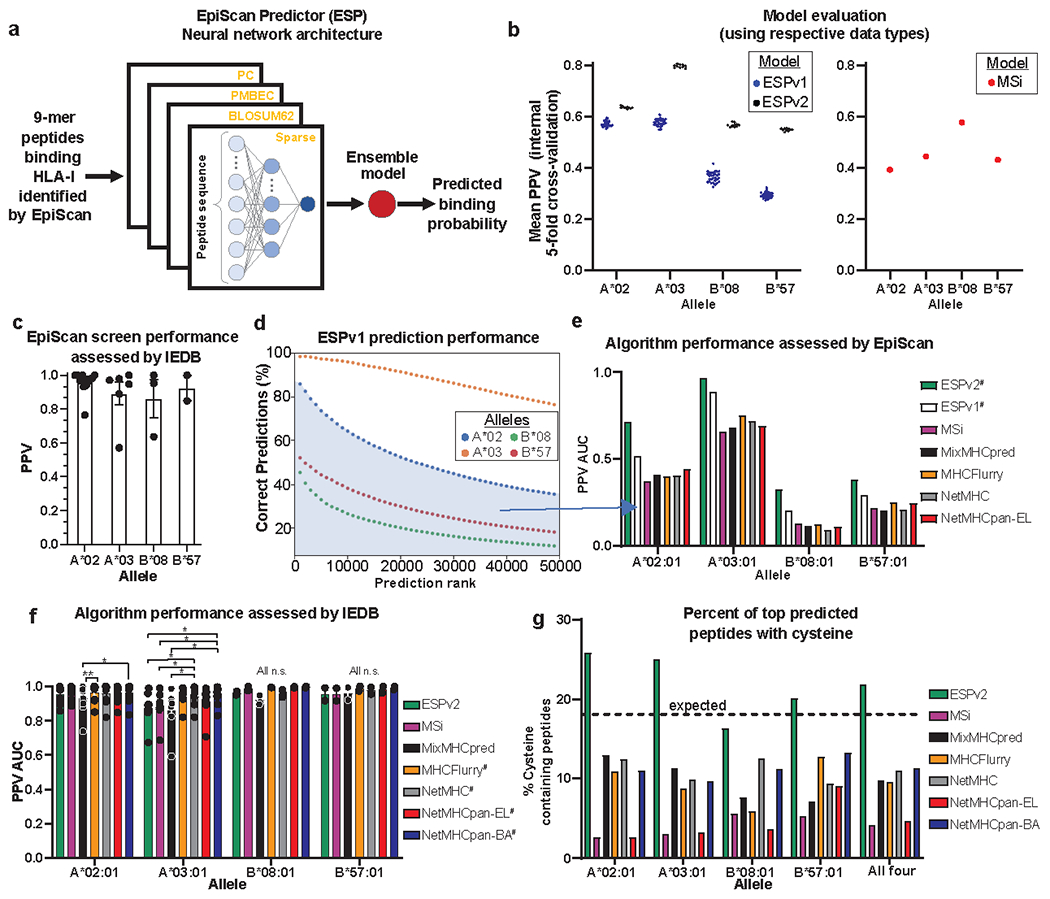
Identification of CD8+ T cell epitopes is critical for the development of immunotherapeutics. Existing methods for major histocompatibility complex class I (MHC class I) ligand discovery are time intensive, specialized and unable to interrogate specific proteins on a large scale. Here, we present EpiScan, which uses surface MHC class I levels as a readout for whether a genetically encoded peptide is an MHC class I ligand. Predetermined starting pools composed of >100,000 peptides can be designed using oligonucleotide synthesis, permitting large-scale MHC class I screening. We exploit this programmability of EpiScan to uncover an unappreciated role for cysteine that increases the number of predicted ligands by 9-21%, reveal affinity hierarchies by analysis of biased anchor peptide libraries and screen viral proteomes for MHC class I ligands. Using these data, we generate and iteratively refine peptide binding predictions to create EpiScan Predictor. EpiScan Predictor performs comparably to other state-of-the-art MHC class I peptide binding prediction algorithms without suffering from underrepresentation of cysteine-containing peptides. Thus, targeted immunopeptidomics using EpiScan will accelerate CD8+ T cell epitope discovery toward the goal of individual-specific immunotherapeutics.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Figures
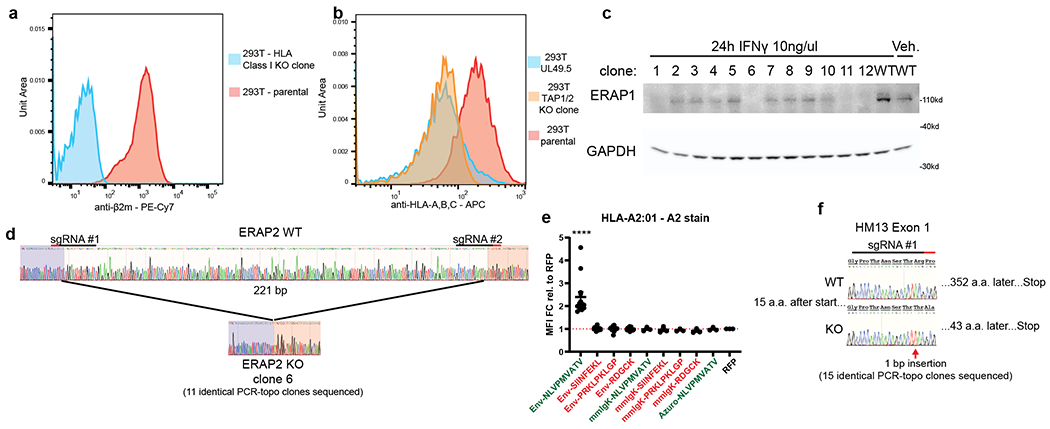
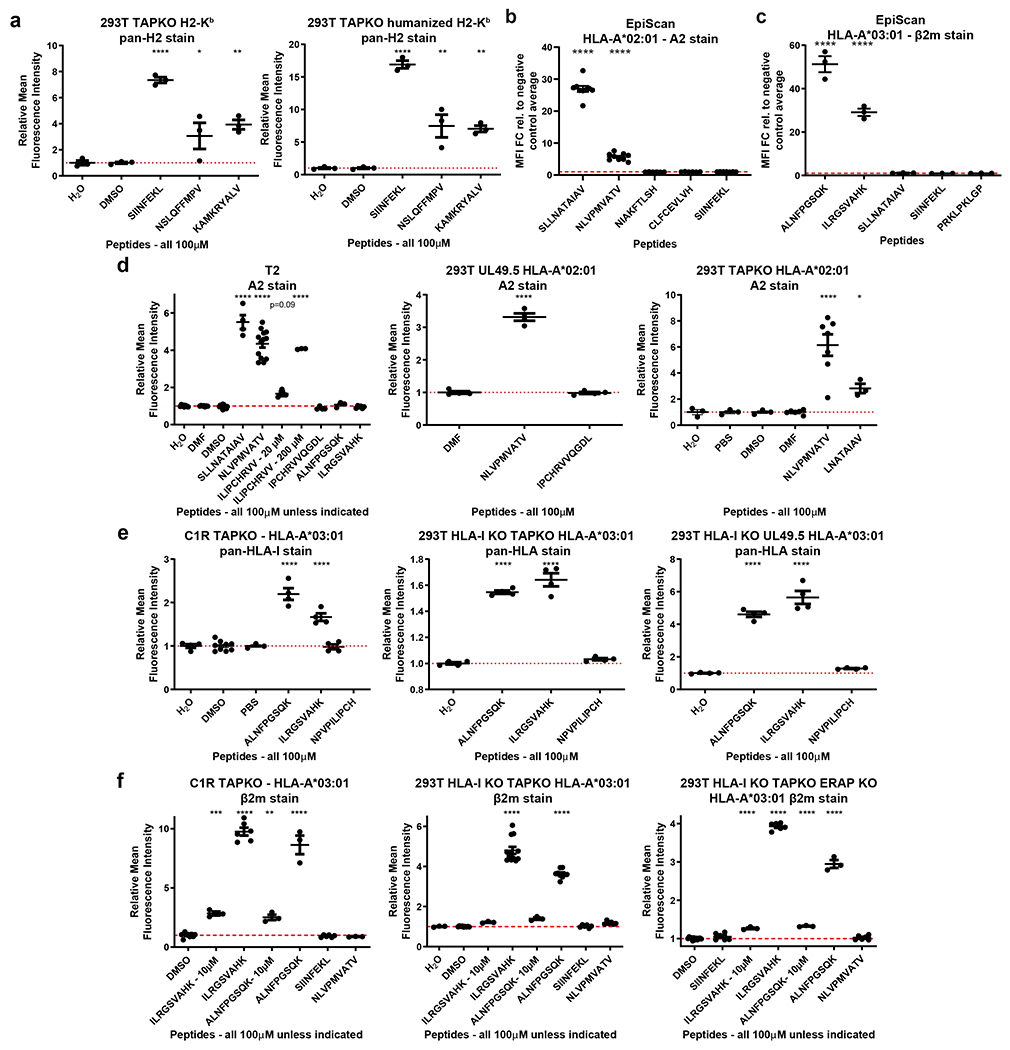
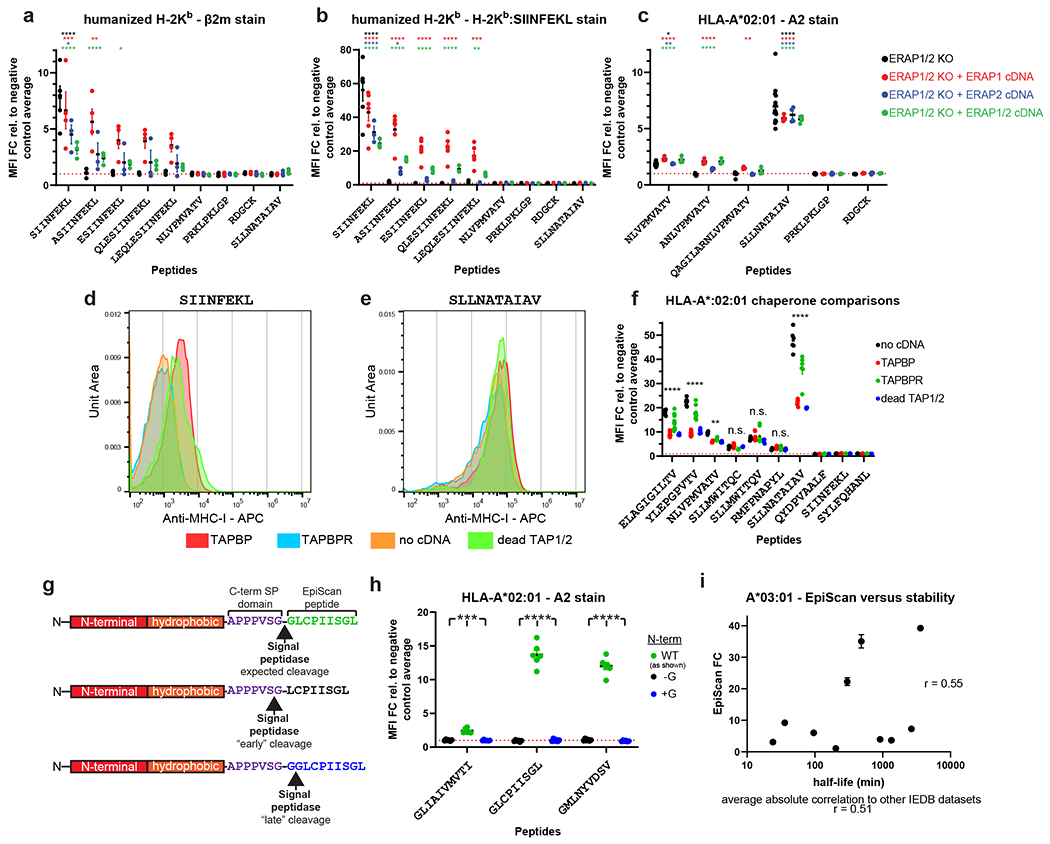
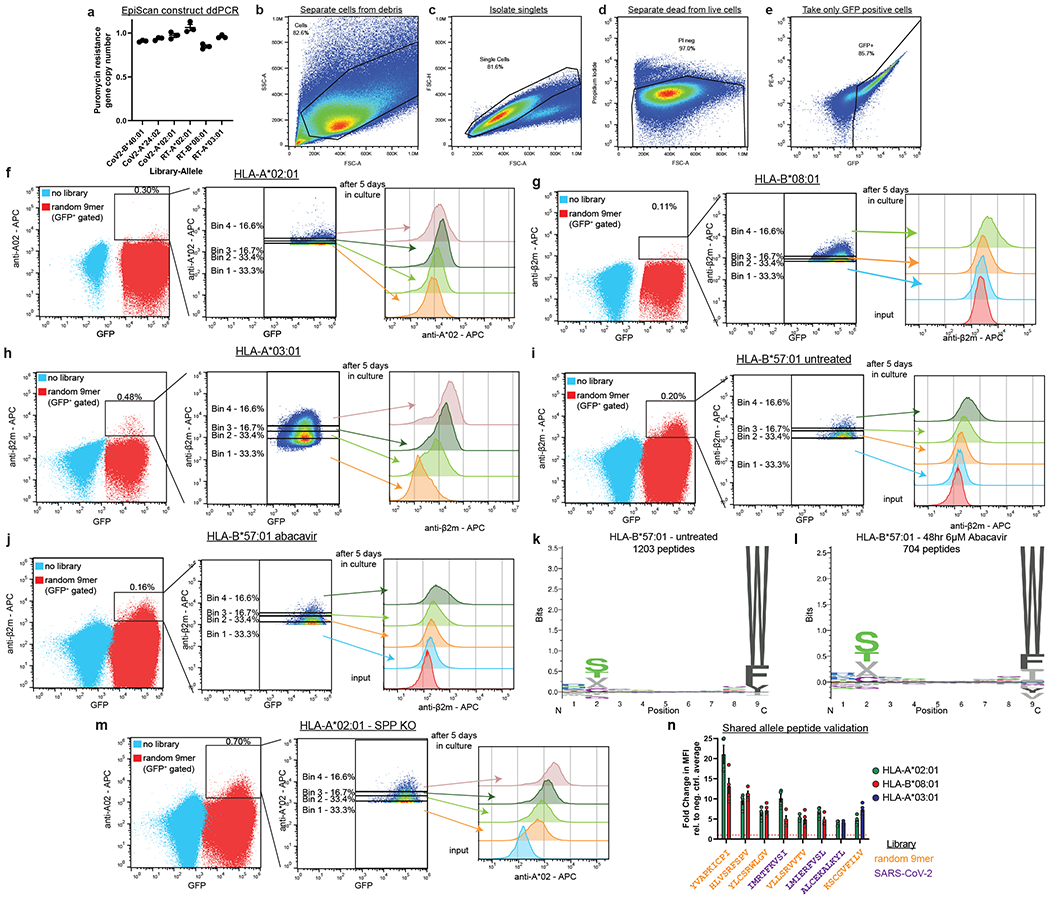
References
-
- Rock KL et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771 (1994). - PubMed
-
- Neefjes J, Jongsma MLM, Paul P & Bakke O Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol 11, (2011). - PubMed
-
- Shen L, Sigal LJ, Boes M & Rock KL Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 21, 155–165 (2004). - PubMed
Methods-only References
-
- Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
