

A Boolean algebra for genetic variants
- PMID: 36594541
- PMCID: PMC9879725
- DOI: 10.1093/bioinformatics/btad001
A Boolean algebra for genetic variants
Abstract
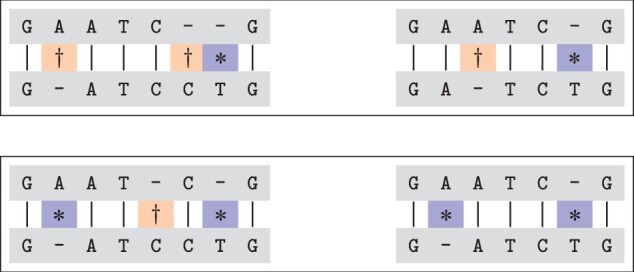
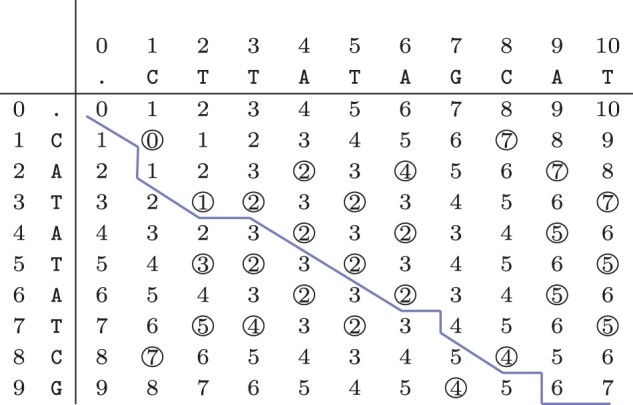
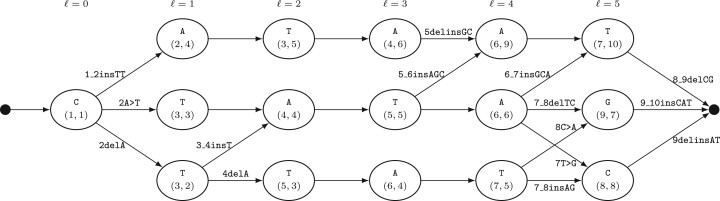
Motivation: Beyond identifying genetic variants, we introduce a set of Boolean relations, which allows for a comprehensive classification of the relations of every pair of variants by taking all minimal alignments into account. We present an efficient algorithm to compute these relations, including a novel way of efficiently computing all minimal alignments within the best theoretical complexity bounds.
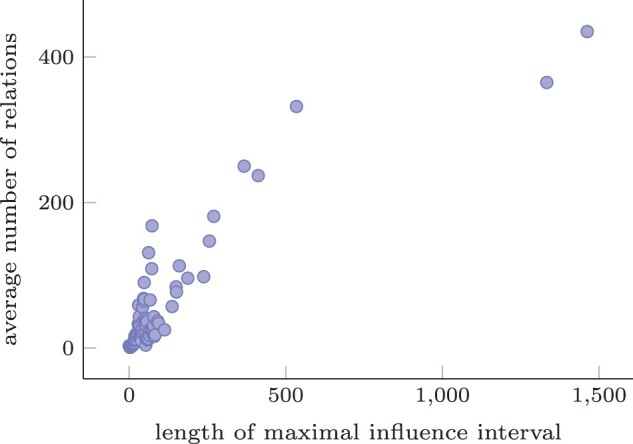
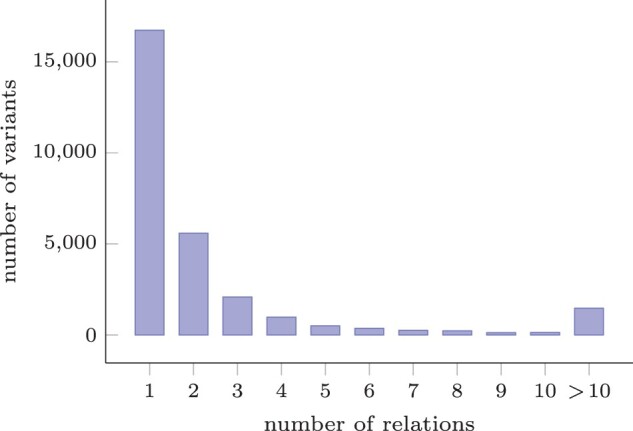
Results: We show that these relations are common, and many non-trivial, for variants of the CFTR gene in dbSNP. Ultimately, we present an approach for the storing and indexing of variants in the context of a database that enables efficient querying for all these relations.
Availability and implementation: A Python implementation is available at https://github.com/mutalyzer/algebra/tree/v0.2.0 as well as an interface at https://mutalyzer.nl/algebra.
© The Author(s) 2023. Published by Oxford University Press.
Figures

References
-
- Allen J.F. (1983) Maintaining knowledge about temporal intervals. Commun. ACM, 26, 832–843.
-
- Backurs A., Indyk P. (2017) Edit distance cannot be computed in strongly subquadratic time (unless SETH is false). arXiv, arXiv:1412.0348, preprint: not peer reviewed. 10.48550/arXiv.1412.0348. - DOI
-
- Bayat A. et al. (2017) Improved VCF normalization for accurate VCF comparison. Bioinformatics, 33, 964–970. - PubMed
