

A biallelic frameshift indel in PPP1R35 as a cause of primary microcephaly
- PMID: 36598158
- PMCID: PMC9928800
- DOI: 10.1002/ajmg.a.63080
A biallelic frameshift indel in PPP1R35 as a cause of primary microcephaly
Abstract
Protein phosphatase 1 regulatory subunit 35 (PPP1R35) encodes a centrosomal protein required for recruiting microtubule-binding elongation machinery. Several proteins in this centriole biogenesis pathway correspond to established primary microcephaly (MCPH) genes, and multiple model organism studies hypothesize PPP1R35 as a candidate MCPH gene. Here, using exome sequencing (ES) and family-based rare variant analyses, we report a homozygous, frameshifting indel deleting the canonical stop codon in the last exon of PPP1R35 [Chr7: c.753_*3delGGAAGCGTAGACCinsCG (p.Trp251Cysfs*22)]; the variant allele maps in a 3.7 Mb block of absence of heterozygosity (AOH) in a proband with severe MCPH (-4.3 SD at birth, -6.1 SD by 42 months), pachygyria, and global developmental delay from a consanguineous Turkish kindred. Droplet digital PCR (ddPCR) confirmed mutant mRNA expression in fibroblasts. In silico prediction of the translation of mutant PPP1R35 is expected to be elongated by 18 amino acids before encountering a downstream stop codon. This complex indel allele is absent in public databases (ClinVar, gnomAD, ARIC, 1000 genomes) and our in-house database of 14,000+ exomes including 1800+ Turkish exomes supporting predicted pathogenicity. Comprehensive literature searches for PPP1R35 variants yielded two probands affected with severe microcephaly (-15 SD and -12 SD) with the same homozygous indel from a single, consanguineous, Iranian family from a cohort of 404 predominantly Iranian families. The lack of heterozygous cases in two large cohorts representative of the genetic background of these two families decreased our suspicion of a founder allele and supports the contention of a recurrent mutation. We propose two potential secondary structure mutagenesis models for the origin of this variant allele mediated by hairpin formation between complementary GC rich segments flanking the stop codon via secondary structure mutagenesis.
Keywords: PPP1R35; centriole; centrosome; complex indel; microcephaly; primary microcephaly.
© 2023 Wiley Periodicals LLC.
Conflict of interest statement
Figures
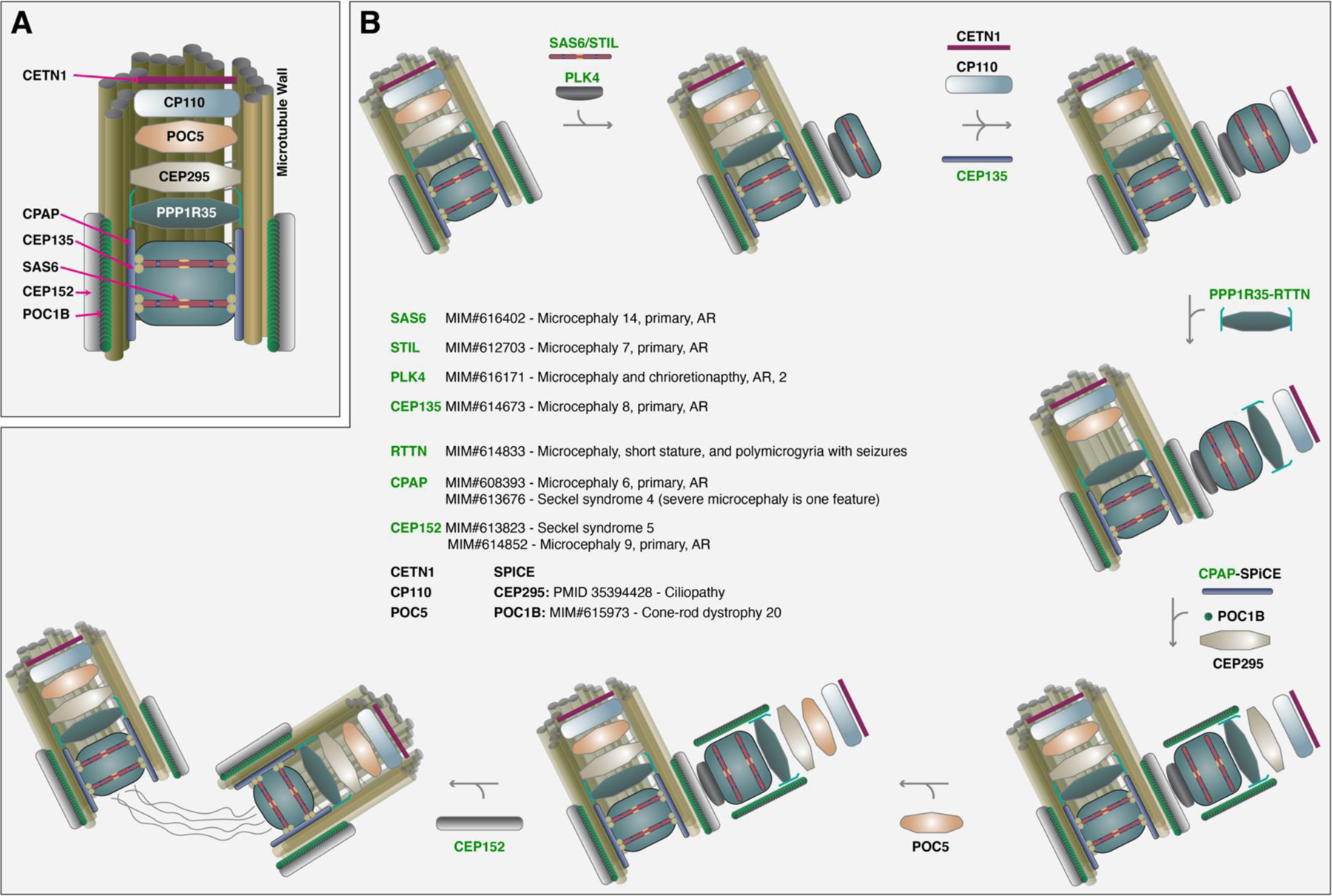
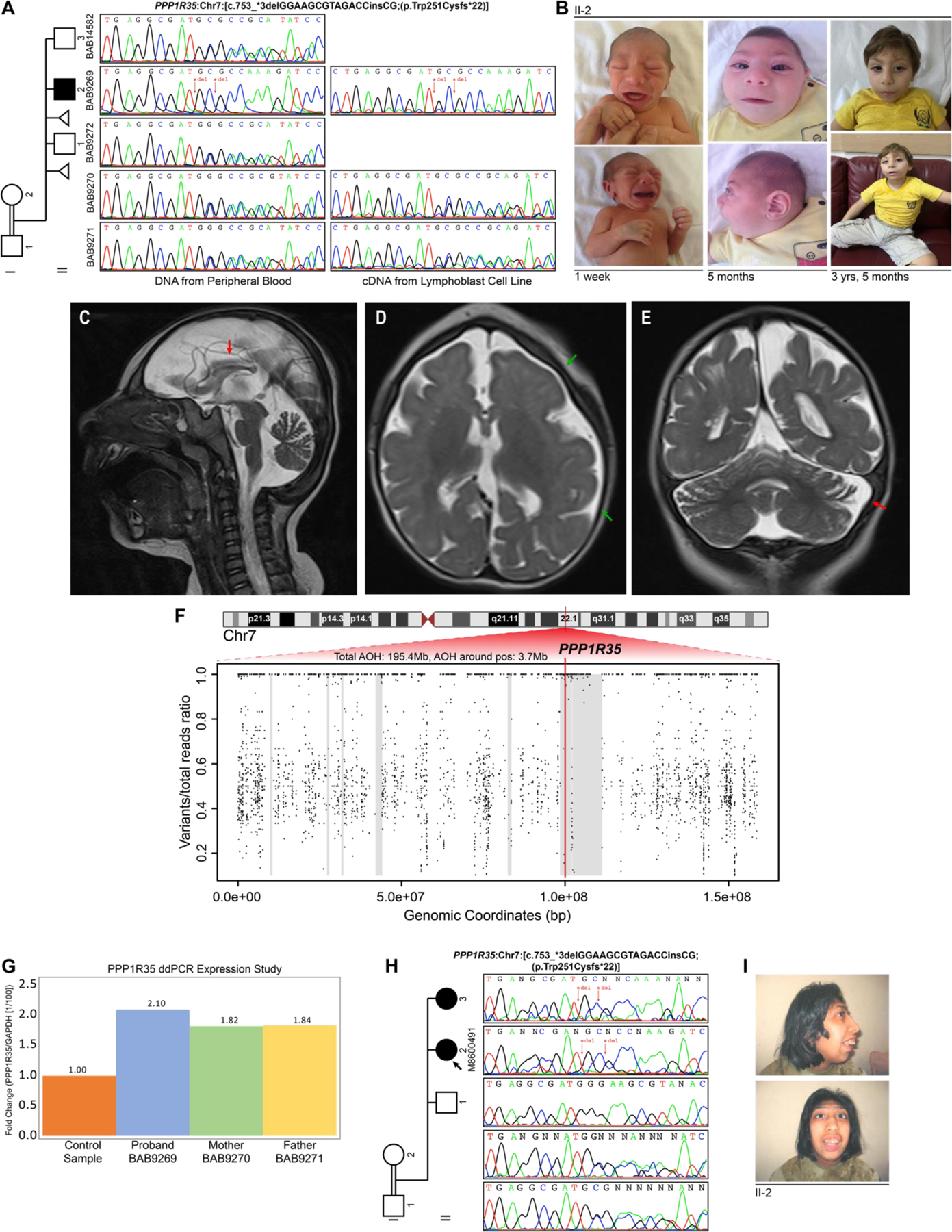
References
-
- Abe S, Katagiri T, Saito-Hisaminato A, Usami S, Inoue Y, Tsunoda T, & Nakamura Y (2003). Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. American Journal of Human Genetics, 72(1), 73–82. 10.1086/345398 - DOI - PMC - PubMed
-
- Archambault D, Cheong A, Iverson E, Tremblay KD, & Mager J (2020). Protein phosphatase 1 regulatory subunit 35 is required for ciliogenesis, notochord morphogenesis, and cell-cycle progression during murine development. Developmental Biology, 465(1), 1–10. 10.1016/j.ydbio.2020.06.011 - DOI - PMC - PubMed
-
- Ashwal S, Michelson D, Plawner L, Dobyns WB, & Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. (2009). Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology, 73(11), 887–897. 10.1212/WNL.0b013e3181b783f7 - DOI - PMC - PubMed
Web Resources
-
- BafCalculator, https://github.com/BCM-Lupskilab/BafCalculator
-
- OMIM, https://www.omim.org/
-
- UniProt, https://www.uniprot.org/
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
