

CACNA1C-Related Channelopathies
- PMID: 36598608
- PMCID: PMC10576998
- DOI: 10.1007/164_2022_624
CACNA1C-Related Channelopathies
Abstract
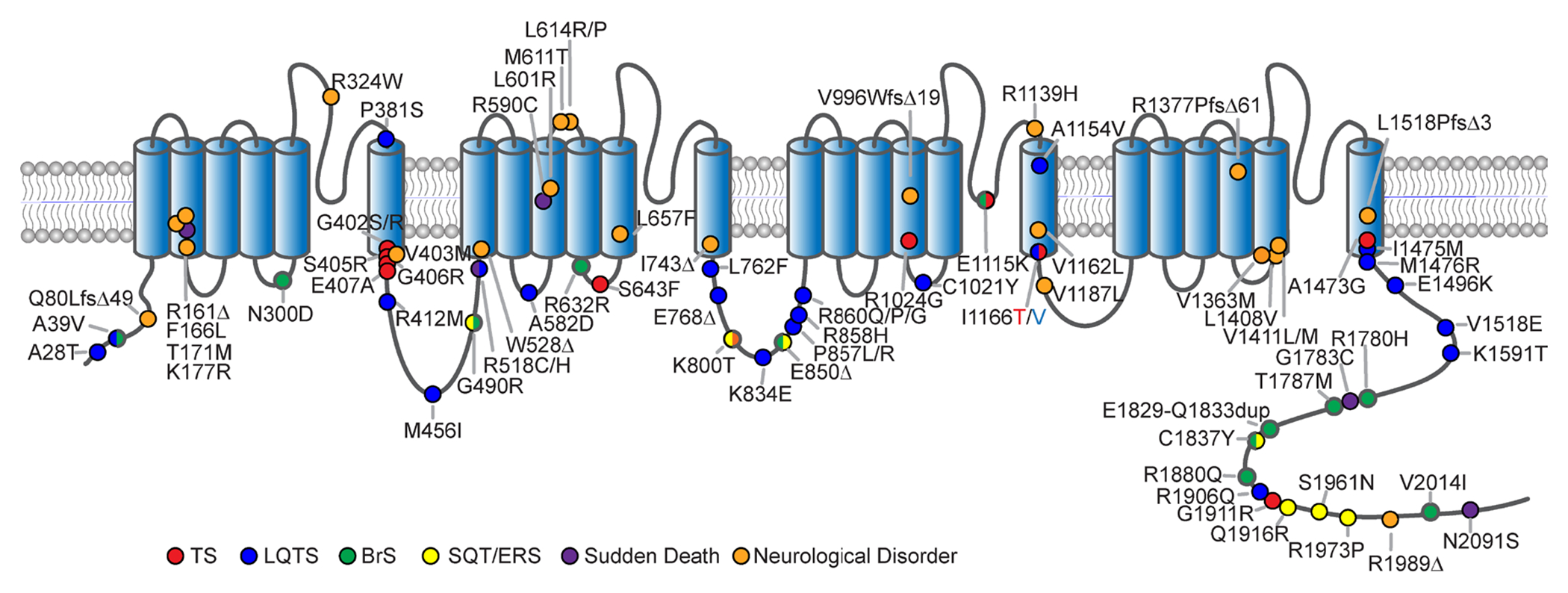
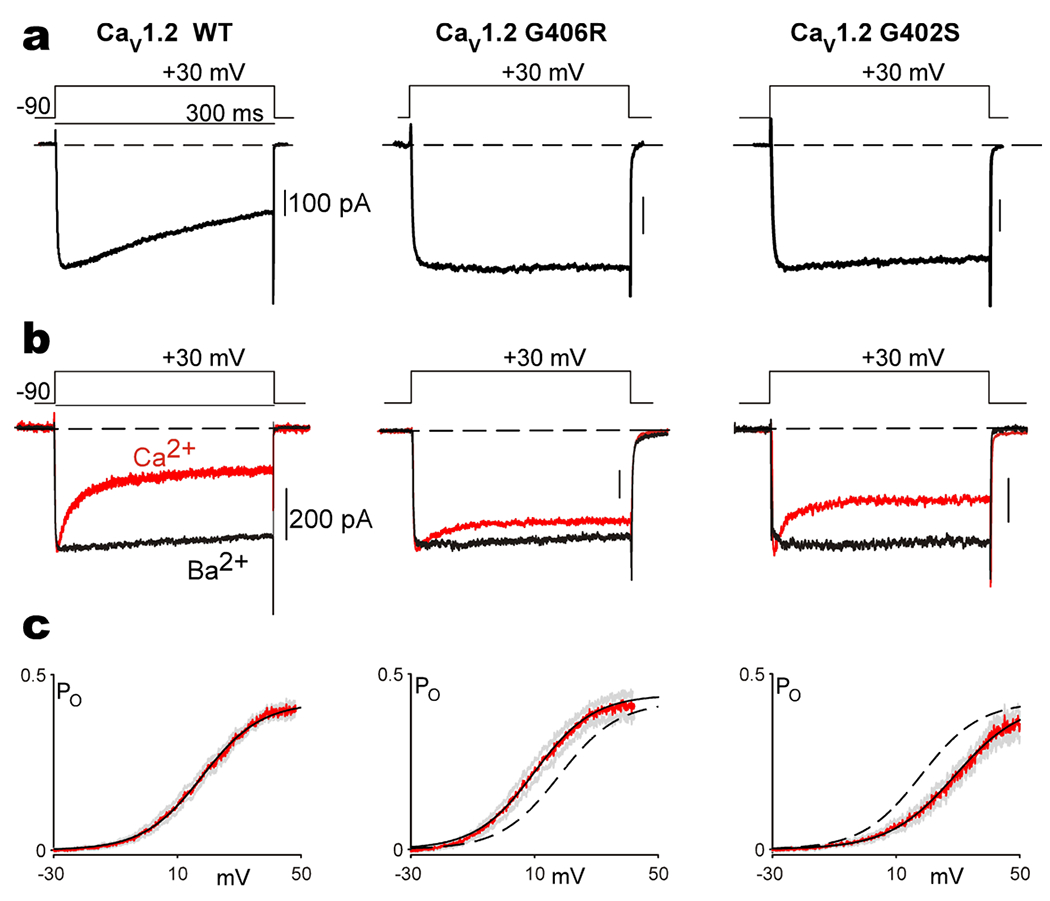
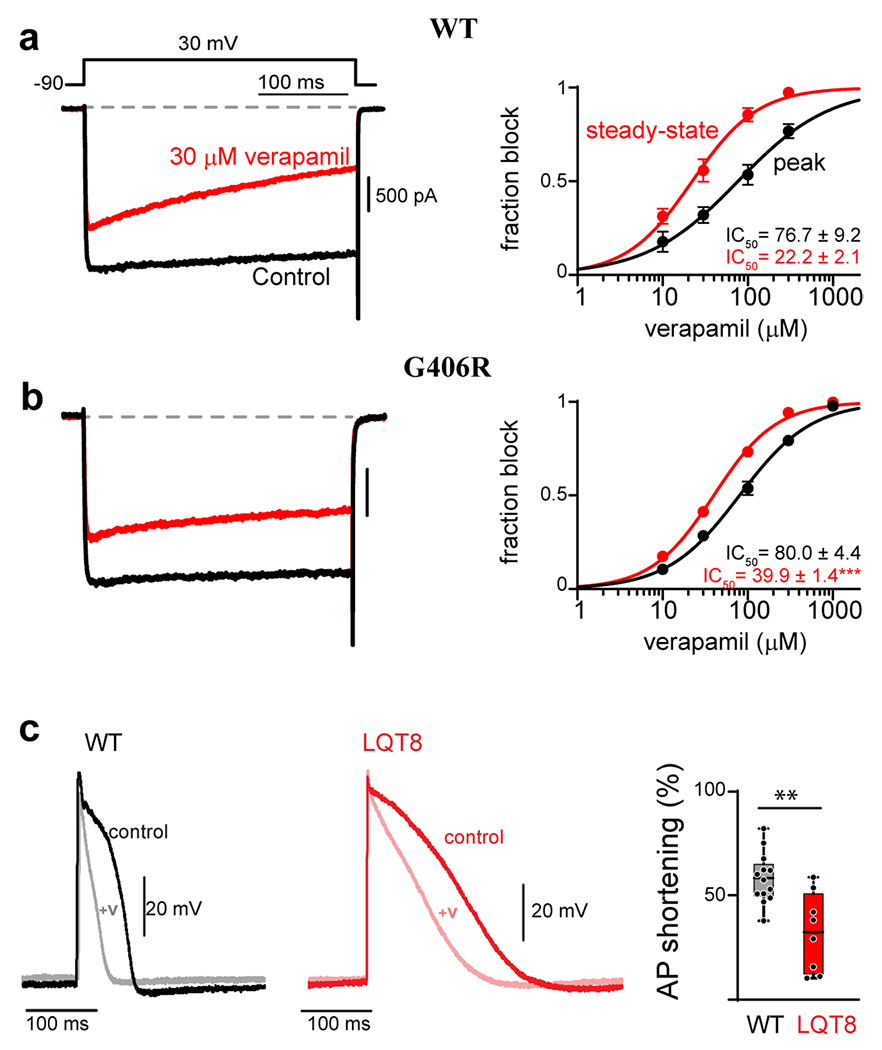
The CACNA1C gene encodes the pore-forming subunit of the CaV1.2 L-type Ca2+ channel, a critical component of membrane physiology in multiple tissues, including the heart, brain, and immune system. As such, mutations altering the function of these channels have the potential to impact a wide array of cellular functions. The first mutations identified within CACNA1C were shown to cause a severe, multisystem disorder known as Timothy syndrome (TS), which is characterized by neurodevelopmental deficits, long-QT syndrome, life-threatening cardiac arrhythmias, craniofacial abnormalities, and immune deficits. Since this initial description, the number and variety of disease-associated mutations identified in CACNA1C have grown tremendously, expanding the range of phenotypes observed in affected patients. CACNA1C channelopathies are now known to encompass multisystem phenotypes as described in TS, as well as more selective phenotypes where patients may exhibit predominantly cardiac or neurological symptoms. Here, we review the impact of genetic mutations on CaV1.2 function and the resultant physiological consequences.
Keywords: CACNA1C; CaV1.2; Channelopathy; L-type calcium channel; Timothy syndrome.
© 2023. The Author(s), under exclusive license to Springer Nature Switzerland AG.
Figures
References
-
- Berger SM, Bartsch D, The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357, 463–476 (2014). - PubMed
-
- Bers DM, Ca regulation in cardiac muscle. Med Sci Sports Exerc 23, 1157–1162 (1991). - PubMed
-
- Lederer WJ et al., Excitation-contraction coupling in heart cells. Roles of the sodium-calcium exchange, the calcium current, and the sarcoplasmic reticulum. Annals of the New York Academy of Sciences 588, 190–206 (1990). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
