

CD8+ T cell-intrinsic IL-6 signaling promotes resistance to anti-PD-L1 immunotherapy
- PMID: 36599350
- PMCID: PMC9873827
- DOI: 10.1016/j.xcrm.2022.100878
CD8+ T cell-intrinsic IL-6 signaling promotes resistance to anti-PD-L1 immunotherapy
Abstract
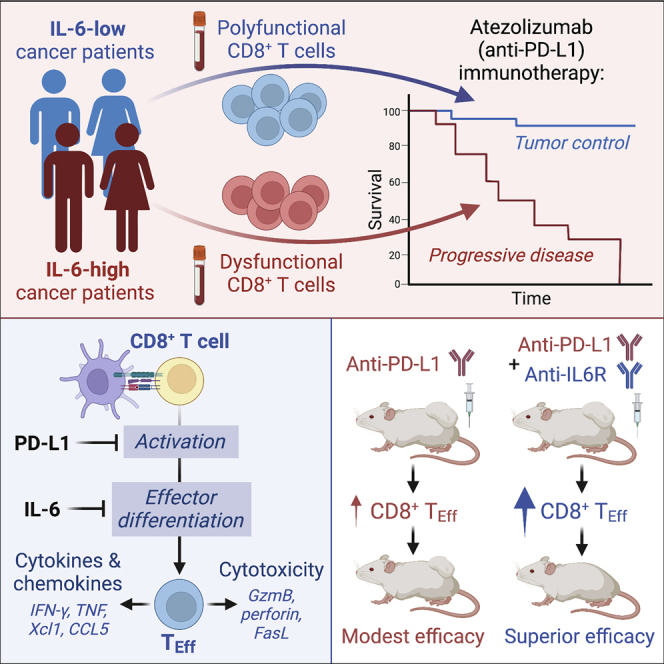
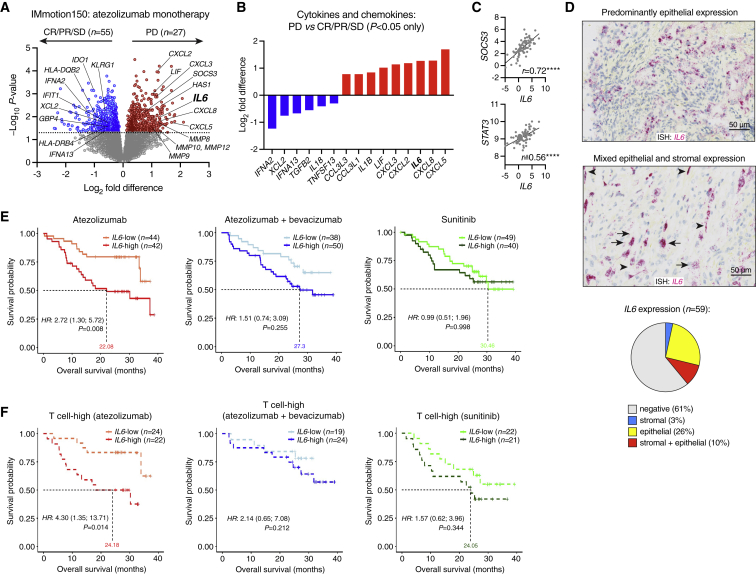
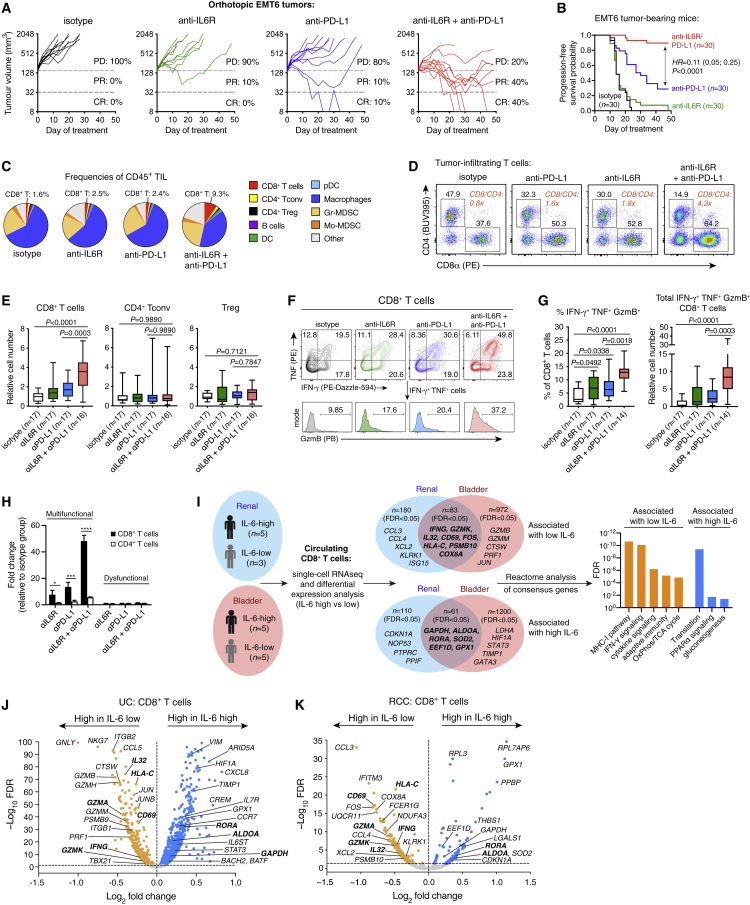
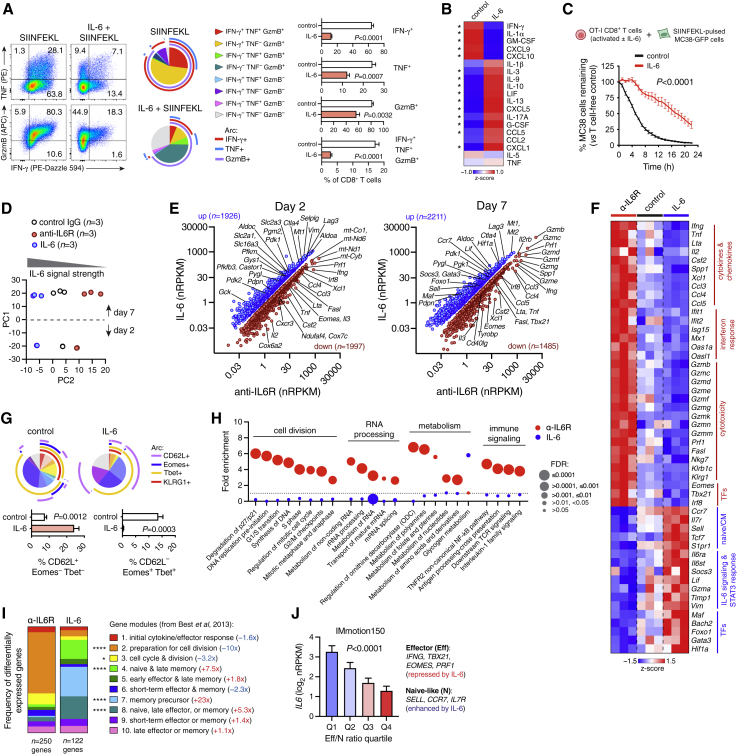
Although immune checkpoint inhibitors (ICIs) are established as effective cancer therapies, overcoming therapeutic resistance remains a critical challenge. Here we identify interleukin 6 (IL-6) as a correlate of poor response to atezolizumab (anti-PD-L1) in large clinical trials of advanced kidney, breast, and bladder cancers. In pre-clinical models, combined blockade of PD-L1 and the IL-6 receptor (IL6R) causes synergistic regression of large established tumors and substantially improves anti-tumor CD8+ cytotoxic T lymphocyte (CTL) responses compared with anti-PD-L1 alone. Circulating CTLs from cancer patients with high plasma IL-6 display a repressed functional profile based on single-cell RNA sequencing, and IL-6-STAT3 signaling inhibits classical cytotoxic differentiation of CTLs in vitro. In tumor-bearing mice, CTL-specific IL6R deficiency is sufficient to improve anti-PD-L1 activity. Thus, based on both clinical and experimental evidence, agents targeting IL-6 signaling are plausible partners for combination with ICIs in cancer patients.
Keywords: CD8 T cell; IL-6; PD-L1; atezolizumab; cancer; checkpoint blockade immunotherapy; clinical trial; interleukin 6.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.A.H., K.Y., L.W., J.E.K., L.L., Y.L., V.G., C.L., D.R., C.O., S.M., S.K., Y.J.C., J.P., Y.S., Z.M., B.B., E.J.F., N.L., H.K., J.Z., M.F., P.W., M.W., I.M., S.J.T., M.M., S.M., L.M., and N.R.W. are employees of Genentech, Inc. M.A.H., K.Y., L.W., J.E.K., L.L., Y.L., P.W., M.M., S.M., L.M., and N.R.W. are inventors on patents related to IL-6. P.S.H. is an employee of Foundation Medicine Inc. K.H. is an employee of Roche Products Ltd. D.F.M. reports a consulting/advisory role for Bristol-Myers Squibb, Merck, Roche/Genentech, Pfizer, Exelixis, Novartis, Eisai, X4 Pharmaceuticals, and Array BioPharma; he also reports that his home institution receives research funding from Prometheus Laboratories. T.P. reports honoraria and consulting/advisory roles with Roche/Genentech, Bristol-Myers Squibb, and Merck; consulting/advisory role with AstraZeneca and Novartis; research funding from AstraZeneca/MedImmune and Roche/Genentech; and other relationships with Ipsen and Bristol-Myers Squibb. L.E. reports honoraria from or consulting/advisory roles with AbbVie, Amgen, AstraZeneca, Bayer, Bristol Meyers Squibb, Celgene, Chugai, eTHeRNA, Genentech, Gritstone, Medimmune, Molecuvax, Macrogenics, Novartis, Peregrine, Replimune, Roche, Silverback, Syndax, and Vaccinex; she reports that her home institution receives funding from Aduro Biotech, AstraZeneca, Breast Cancer Research Foundation, Bristol Meyers Squibb, Corvus, Department of Defense, EMD Serono, Genentech, HeritX, Inc., Maxcyte, Merck, National Cancer Institute, NSABP Foundation, Roche, Tempest, Translational Breast Cancer Research Consortium. J.E.R. has received non-financial support from Roche Genentech and consulting fees from Agensys, Eli Lilly, Sanofi, and Oncogene.
Figures
Comment in
-
Illuminating the mechanism of IL-6-mediated immunotherapy resistance.Cell Rep Med. 2023 Jan 17;4(1):100901. doi: 10.1016/j.xcrm.2022.100901. Cell Rep Med. 2023. PMID: 36652910 Free PMC article.
References
-
- Tsukamoto H., Fujieda K., Miyashita A., Fukushima S., Ikeda T., Kubo Y., Senju S., Ihn H., Nishimura Y., Oshiumi H. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 2018;78:5011–5022. doi: 10.1158/0008-5472.CAN-18-0118. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
