

Influenza vaccination reveals sex dimorphic imprints of prior mild COVID-19
- PMID: 36599369
- PMCID: PMC10481789
- DOI: 10.1038/s41586-022-05670-5
Influenza vaccination reveals sex dimorphic imprints of prior mild COVID-19
Abstract
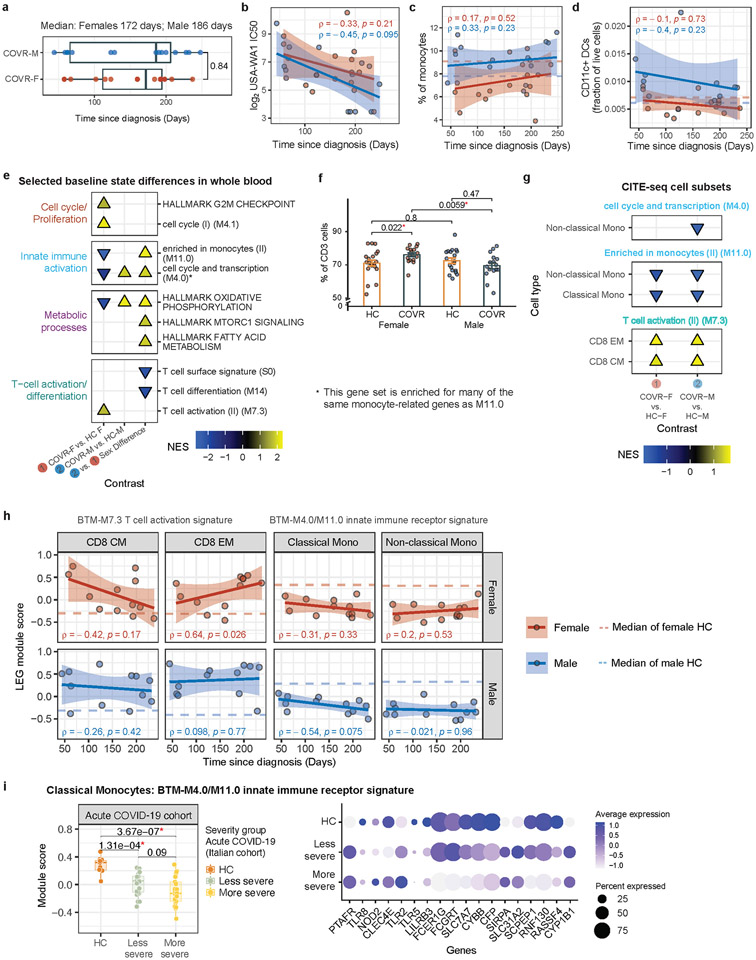
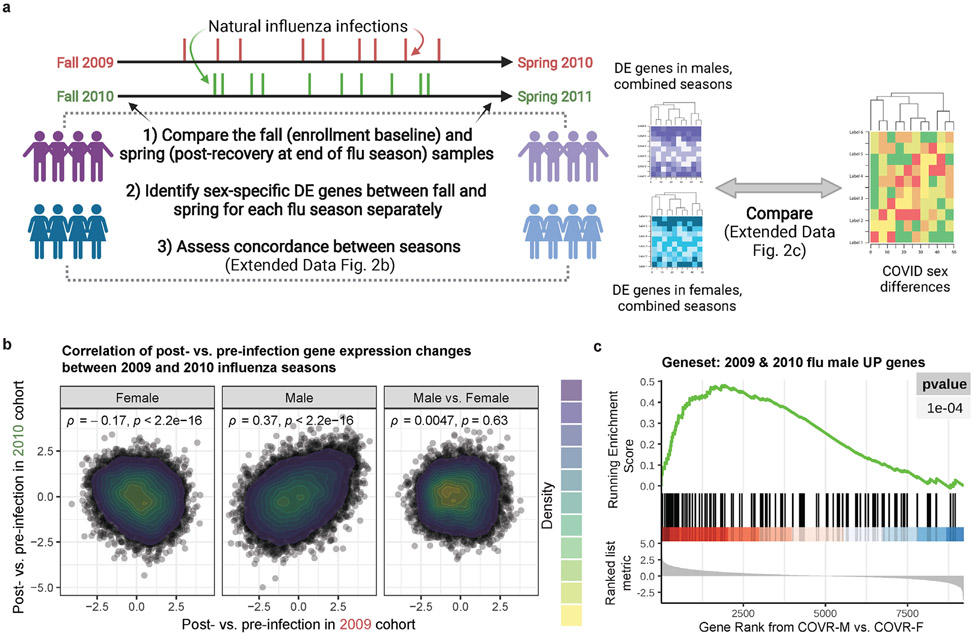
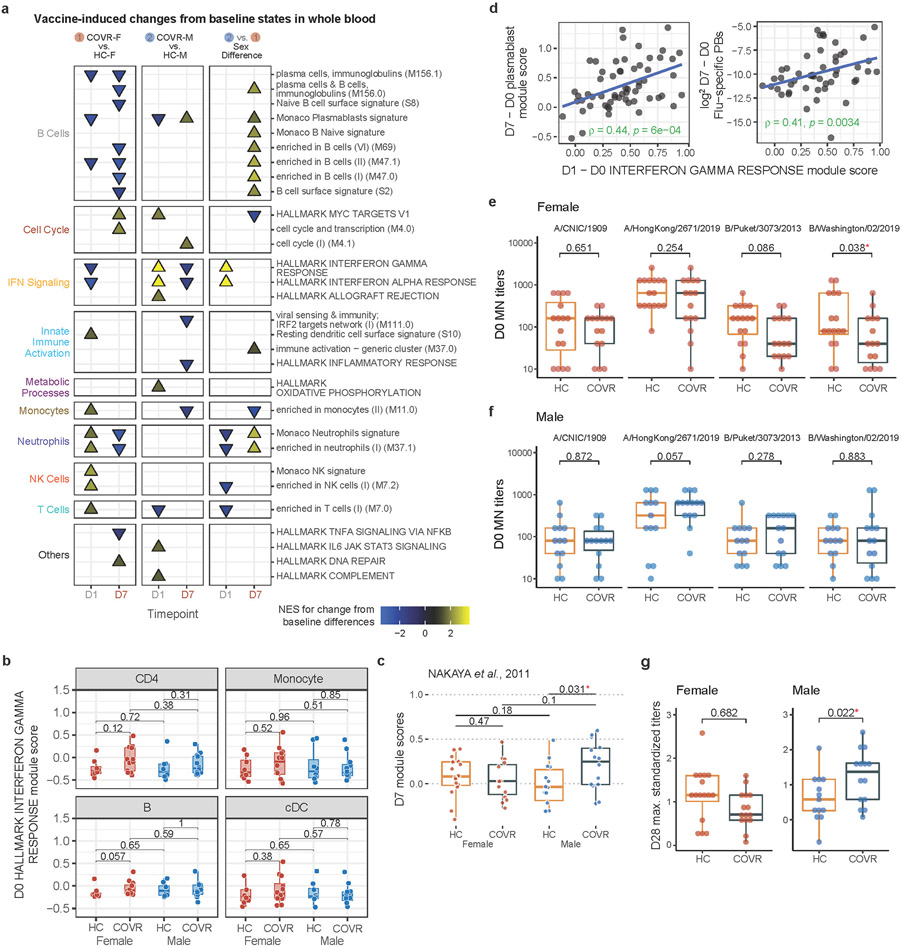
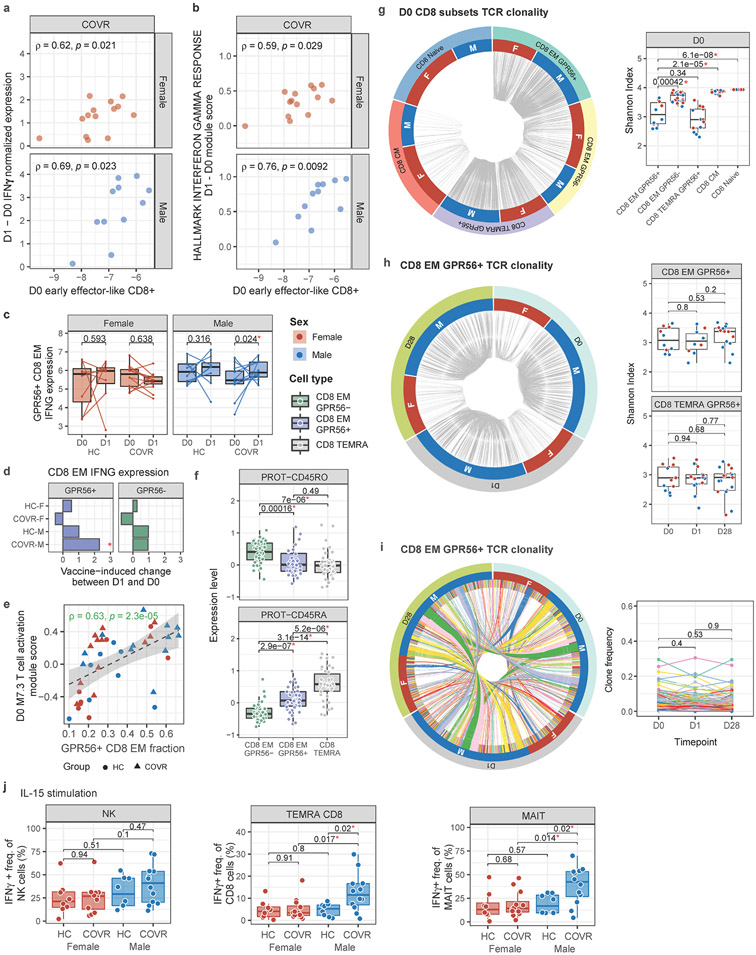
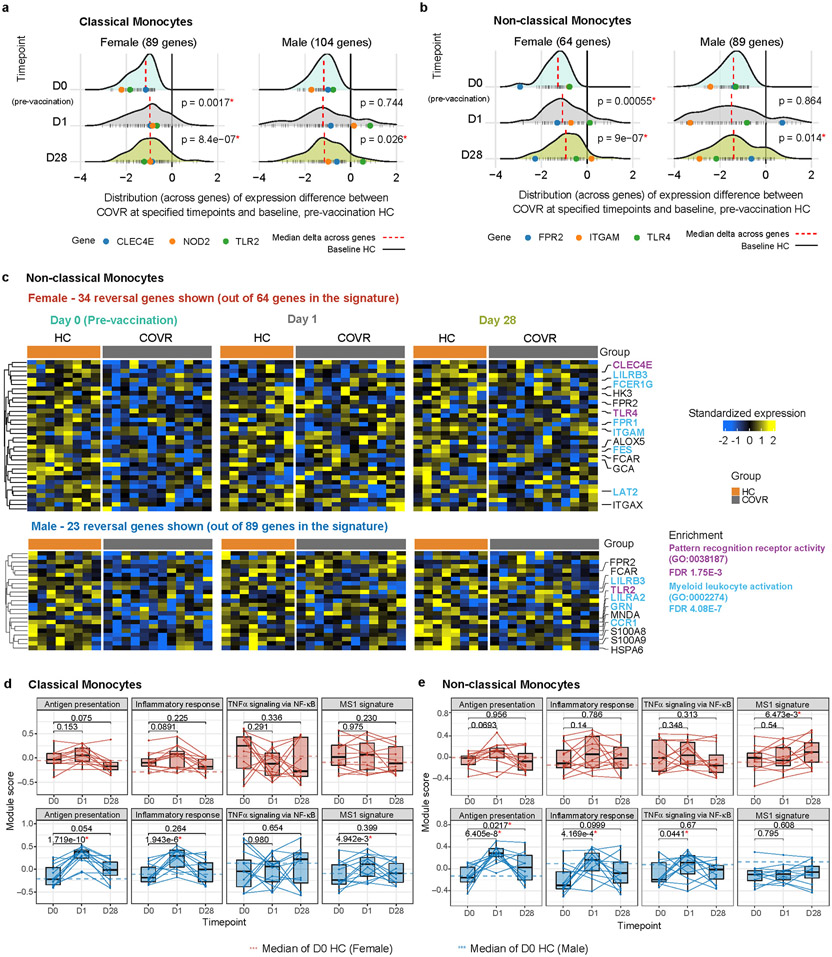
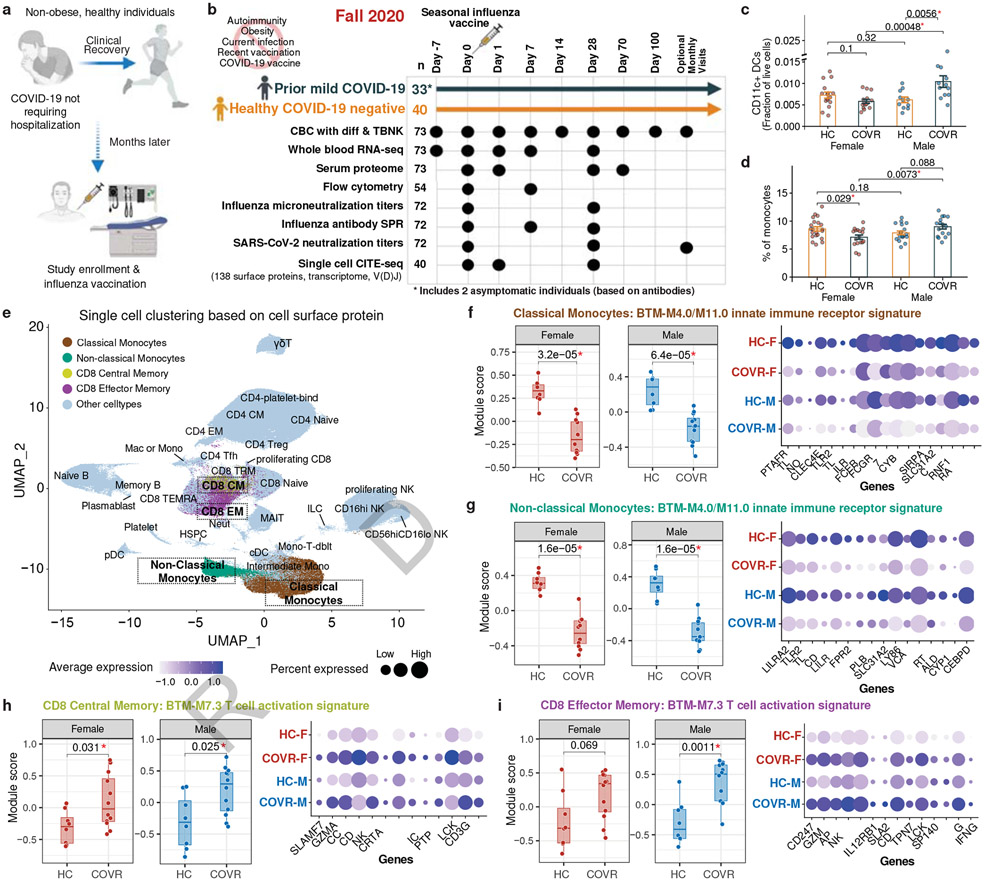
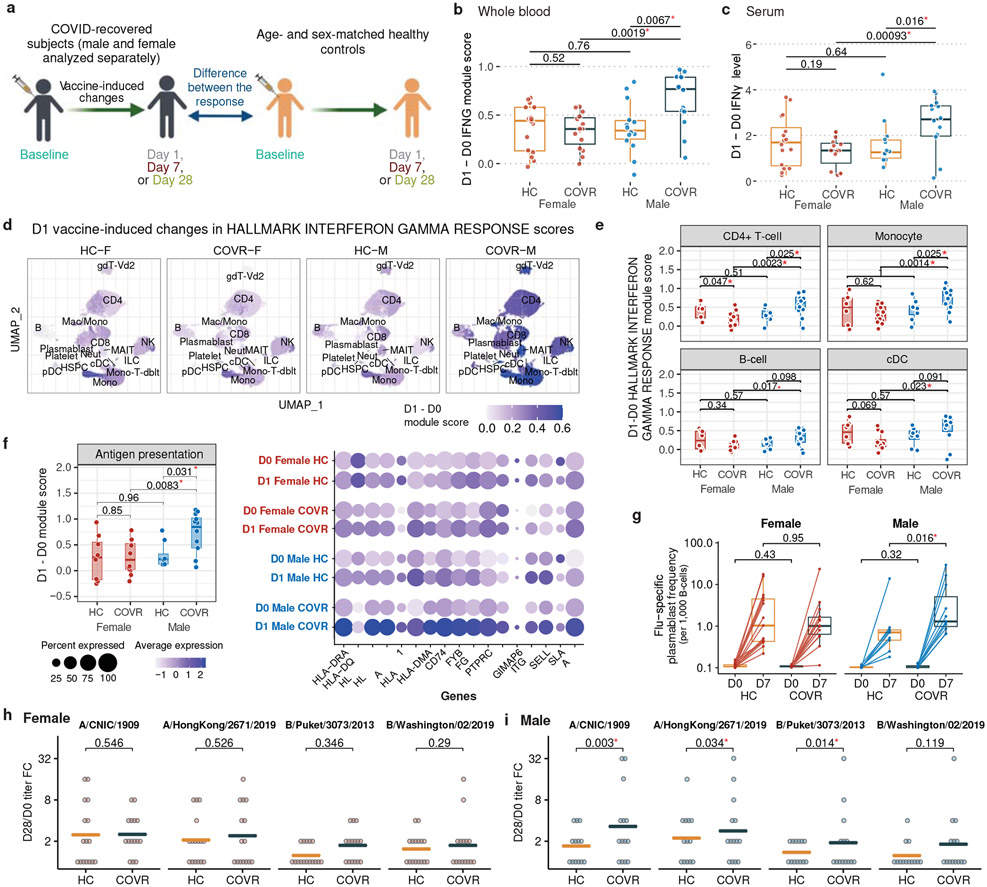
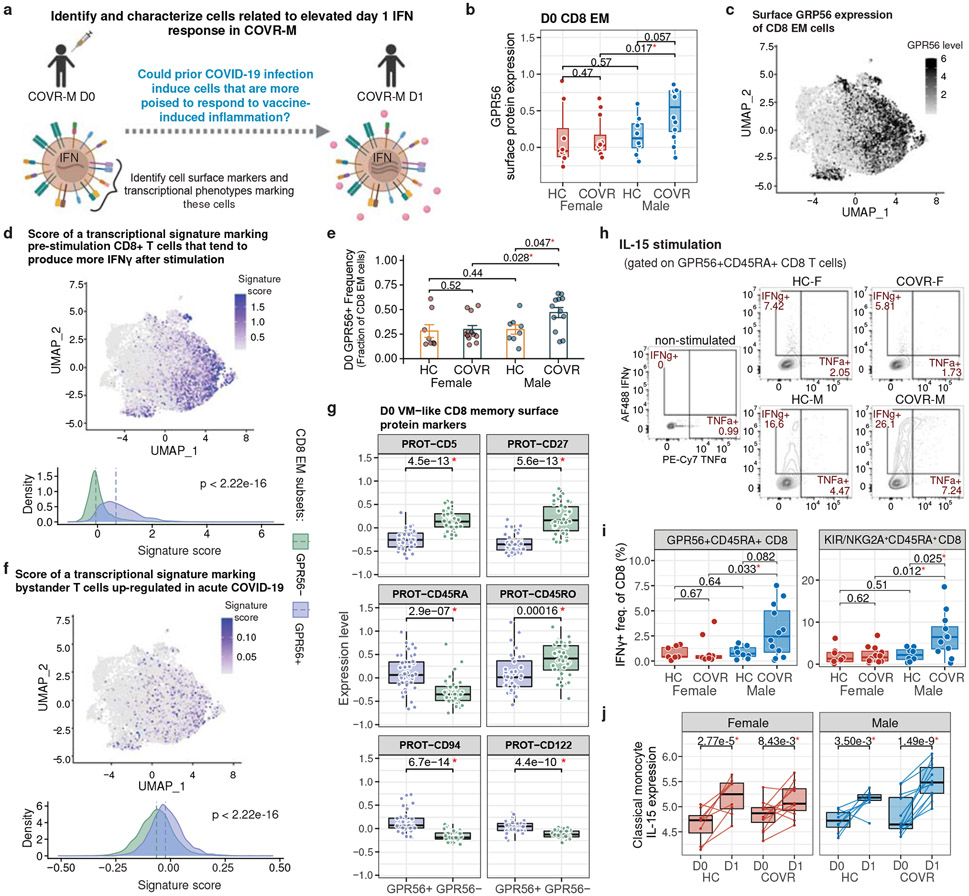
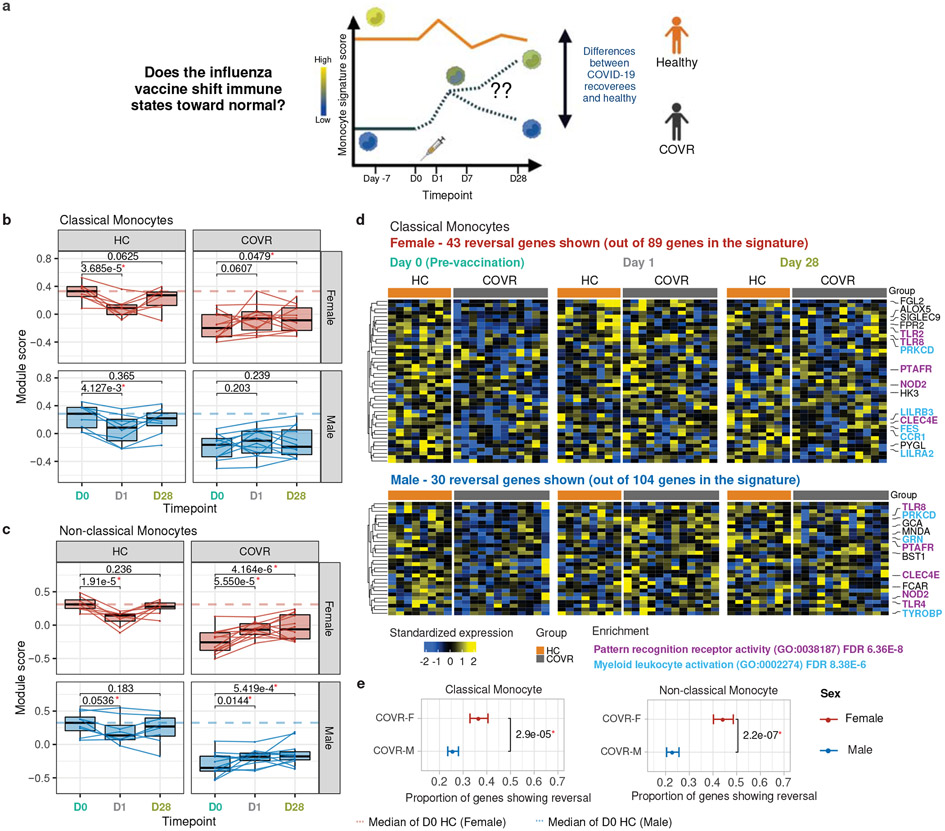
Acute viral infections can have durable functional impacts on the immune system long after recovery, but how they affect homeostatic immune states and responses to future perturbations remain poorly understood1-4. Here we use systems immunology approaches, including longitudinal multimodal single-cell analysis (surface proteins, transcriptome and V(D)J sequences) to comparatively assess baseline immune statuses and responses to influenza vaccination in 33 healthy individuals after recovery from mild, non-hospitalized COVID-19 (mean, 151 days after diagnosis) and 40 age- and sex-matched control individuals who had never had COVID-19. At the baseline and independent of time after COVID-19, recoverees had elevated T cell activation signatures and lower expression of innate immune genes including Toll-like receptors in monocytes. Male individuals who had recovered from COVID-19 had coordinately higher innate, influenza-specific plasmablast, and antibody responses after vaccination compared with healthy male individuals and female individuals who had recovered from COVID-19, in part because male recoverees had monocytes with higher IL-15 responses early after vaccination coupled with elevated prevaccination frequencies of 'virtual memory'-like CD8+ T cells poised to produce more IFNγ after IL-15 stimulation. Moreover, the expression of the repressed innate immune genes in monocytes increased by day 1 to day 28 after vaccination in recoverees, therefore moving towards the prevaccination baseline of the healthy control individuals. By contrast, these genes decreased on day 1 and returned to the baseline by day 28 in the control individuals. Our study reveals sex-dimorphic effects of previous mild COVID-19 and suggests that viral infections in humans can establish new immunological set-points that affect future immune responses in an antigen-agnostic manner.
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Figures
Update of
-
Influenza vaccination and single cell multiomics reveal sex dimorphic immune imprints of prior mild COVID-19.medRxiv [Preprint]. 2022 Sep 1:2022.02.17.22271138. doi: 10.1101/2022.02.17.22271138. medRxiv. 2022. Update in: Nature. 2023 Feb;614(7949):752-761. doi: 10.1038/s41586-022-05670-5. PMID: 35233581 Free PMC article. Updated. Preprint.
References
Main Text References
-
- Tomalka JA, Suthar MS, Diamond MS & Sekaly RP Innate antiviral immunity: how prior exposures can guide future responses. Trends Immunol. 43, 696–705 (2022). - PubMed
Methods and Extended Data Figure References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
