

Integrated intracellular organization and its variations in human iPS cells
- PMID: 36599983
- PMCID: PMC9834050
- DOI: 10.1038/s41586-022-05563-7
Integrated intracellular organization and its variations in human iPS cells
Abstract
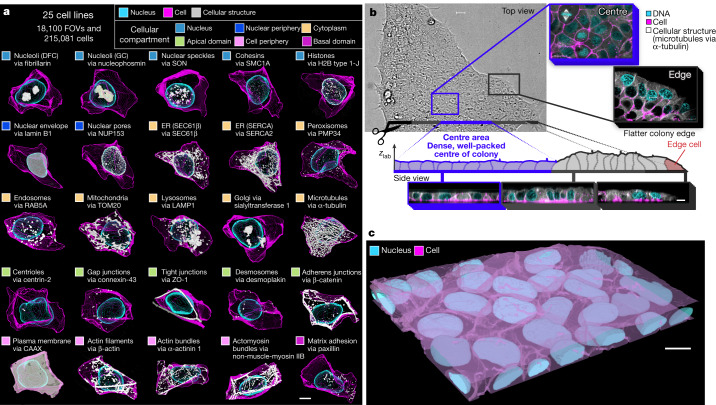
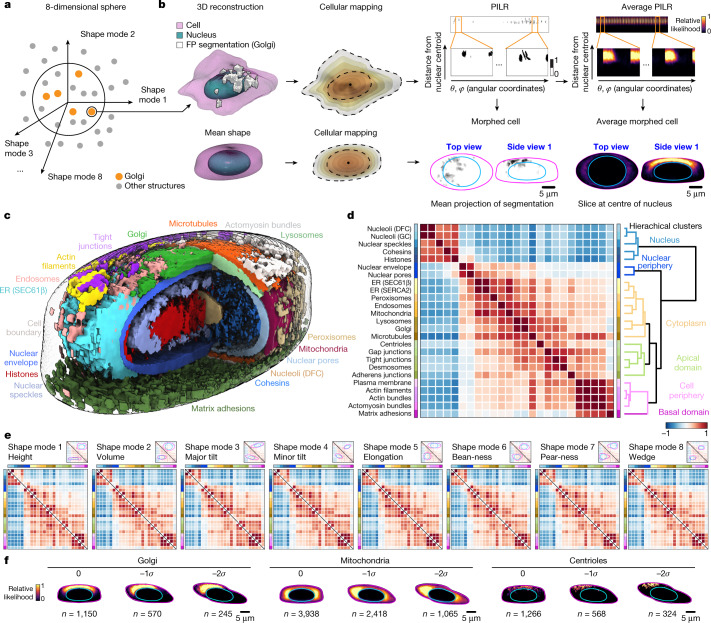
Understanding how a subset of expressed genes dictates cellular phenotype is a considerable challenge owing to the large numbers of molecules involved, their combinatorics and the plethora of cellular behaviours that they determine1,2. Here we reduced this complexity by focusing on cellular organization-a key readout and driver of cell behaviour3,4-at the level of major cellular structures that represent distinct organelles and functional machines, and generated the WTC-11 hiPSC Single-Cell Image Dataset v1, which contains more than 200,000 live cells in 3D, spanning 25 key cellular structures. The scale and quality of this dataset permitted the creation of a generalizable analysis framework to convert raw image data of cells and their structures into dimensionally reduced, quantitative measurements that can be interpreted by humans, and to facilitate data exploration. This framework embraces the vast cell-to-cell variability that is observed within a normal population, facilitates the integration of cell-by-cell structural data and allows quantitative analyses of distinct, separable aspects of organization within and across different cell populations. We found that the integrated intracellular organization of interphase cells was robust to the wide range of variation in cell shape in the population; that the average locations of some structures became polarized in cells at the edges of colonies while maintaining the 'wiring' of their interactions with other structures; and that, by contrast, changes in the location of structures during early mitotic reorganization were accompanied by changes in their wiring.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures















References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
