

Molecular-dynamics simulation methods for macromolecular crystallography
- PMID: 36601807
- PMCID: PMC9815100
- DOI: 10.1107/S2059798322011871
Molecular-dynamics simulation methods for macromolecular crystallography
Abstract
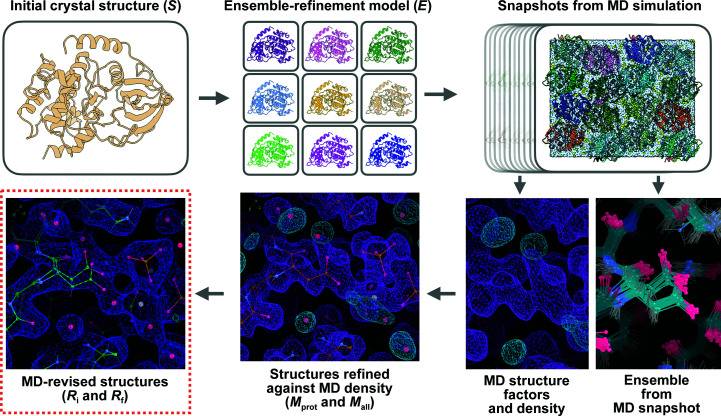
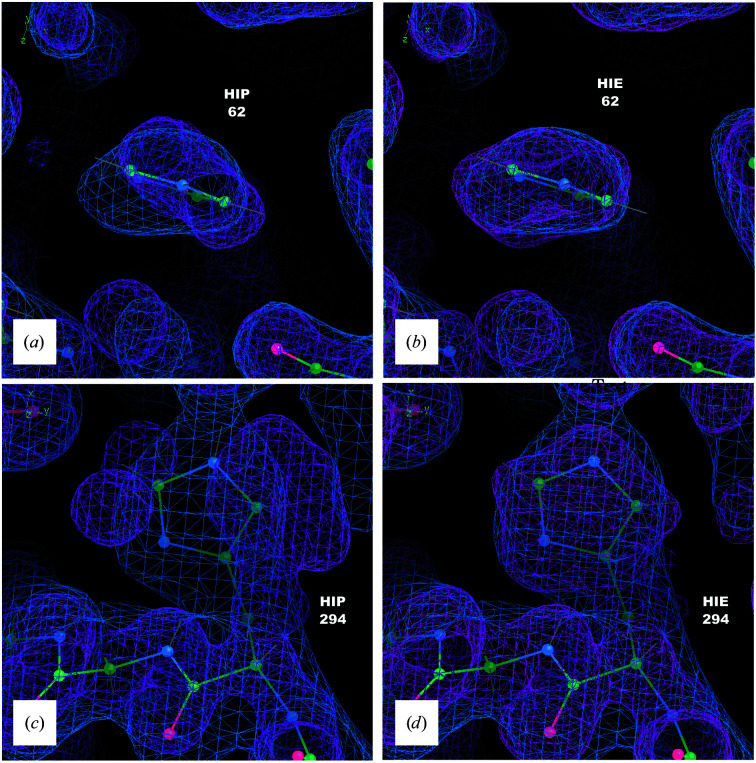
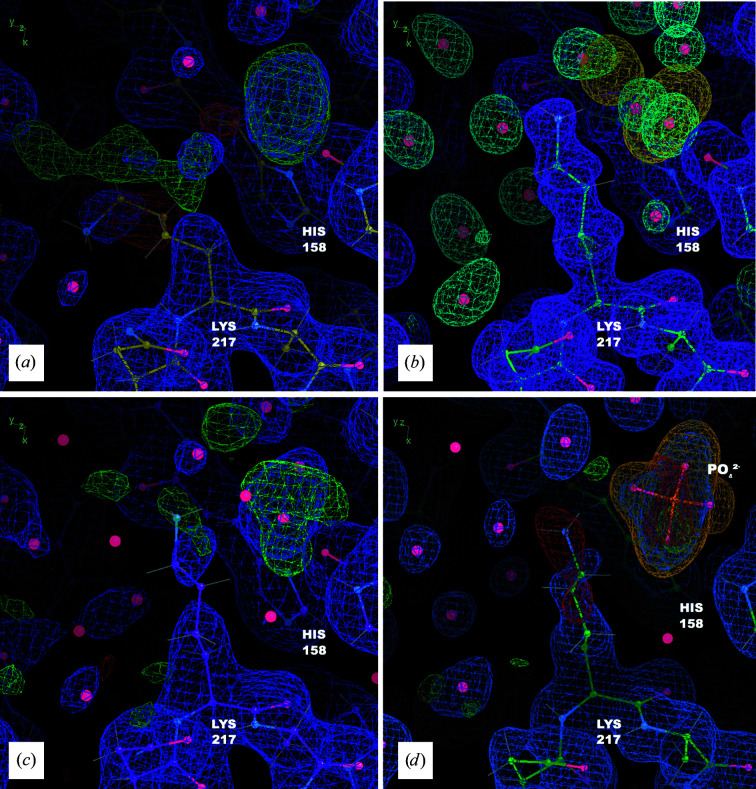
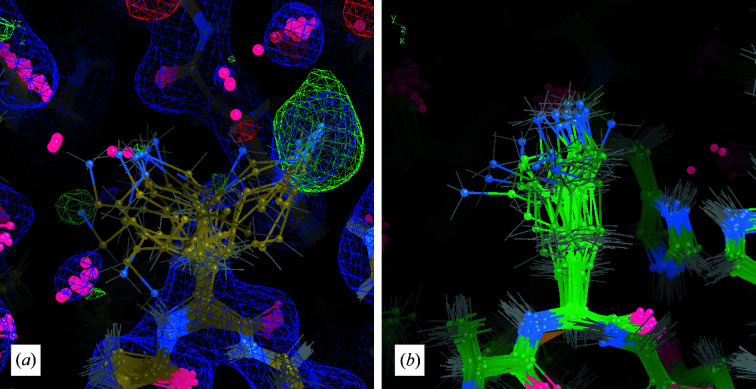
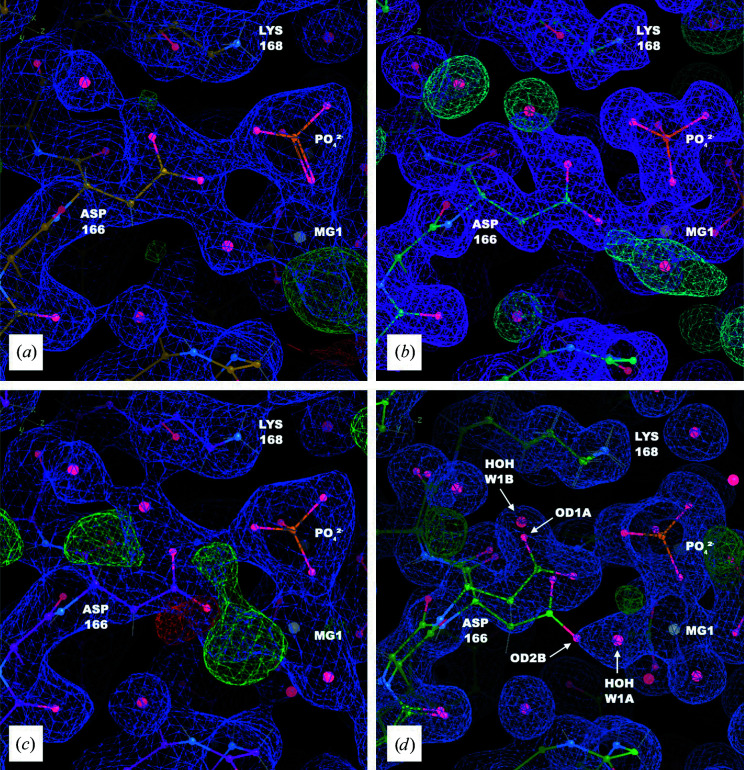
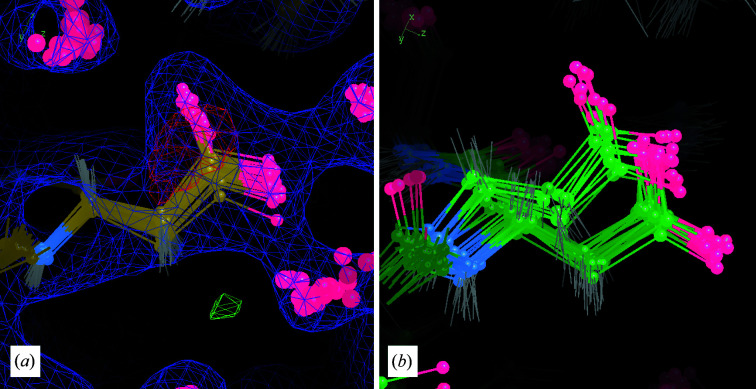
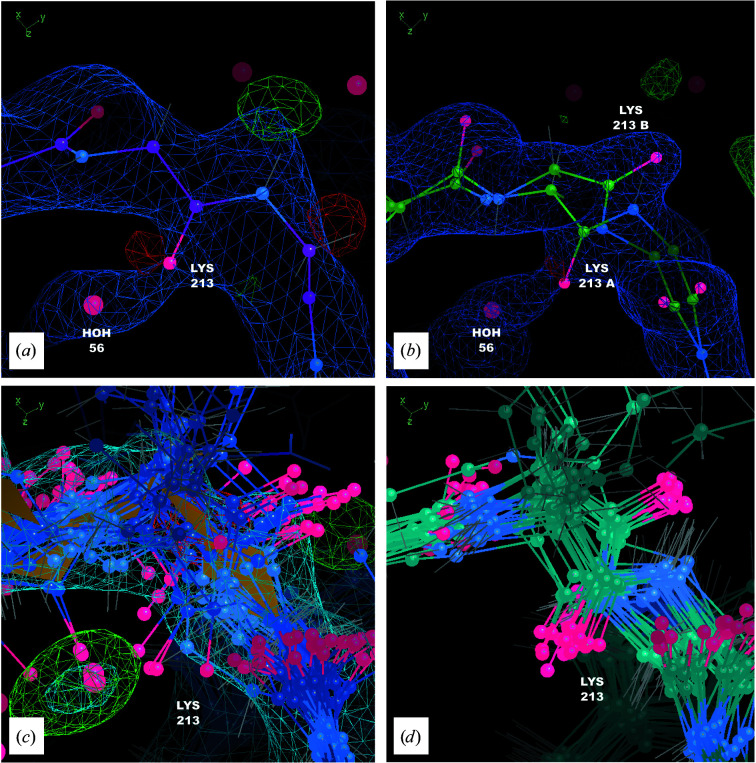
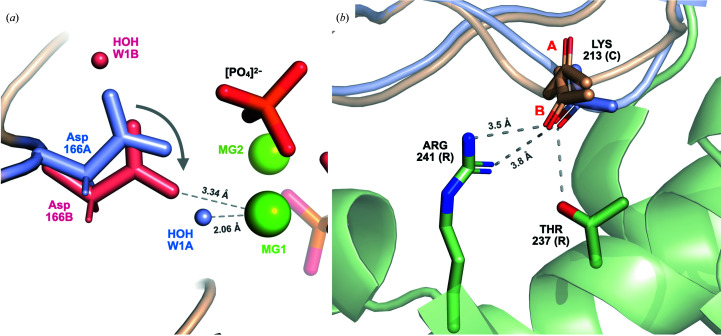
It is investigated whether molecular-dynamics (MD) simulations can be used to enhance macromolecular crystallography (MX) studies. Historically, protein crystal structures have been described using a single set of atomic coordinates. Because conformational variation is important for protein function, researchers now often build models that contain multiple structures. Methods for building such models can fail, however, in regions where the crystallographic density is difficult to interpret, for example at the protein-solvent interface. To address this limitation, a set of MD-MX methods that combine MD simulations of protein crystals with conventional modeling and refinement tools have been developed. In an application to a cyclic adenosine monophosphate-dependent protein kinase at room temperature, the procedure improved the interpretation of ambiguous density, yielding an alternative water model and a revised protein model including multiple conformations. The revised model provides mechanistic insights into the catalytic and regulatory interactions of the enzyme. The same methods may be used in other MX studies to seek mechanistic insights.
Keywords: conformational ensembles; molecular-dynamics simulations; protein kinases; water structure.
open access.
Figures
References
-
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B. & Lindahl, E. (2015). SoftwareX, 1–2, 19–25.
-
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. - PMC - PubMed
MeSH terms
Substances
Grants and funding
- GM108889/NH/NIH HHS/United States
- R01 GM108889/GM/NIGMS NIH HHS/United States
- GM124270/NH/NIH HHS/United States
- GM123159/NH/NIH HHS/United States
- GM124149/NH/NIH HHS/United States
- GM132386/NH/NIH HHS/United States
- R01 GM132386/GM/NIGMS NIH HHS/United States
- R35 GM130389/GM/NIGMS NIH HHS/United States
- R01 GM123159/GM/NIGMS NIH HHS/United States
- R01 GM124149/GM/NIGMS NIH HHS/United States
- R01 GM124270/GM/NIGMS NIH HHS/United States
- T32 CA009523/CA/NCI NIH HHS/United States
- GM130389/NH/NIH HHS/United States
- T32 CA009523/CA/NCI/NH/NIH HHS/United States
