

Dynamics of Microbial Community and Potential Microbial Pollutants in Shopping Malls
- PMID: 36602317
- PMCID: PMC9948725
- DOI: 10.1128/msystems.00576-22
Dynamics of Microbial Community and Potential Microbial Pollutants in Shopping Malls
Abstract
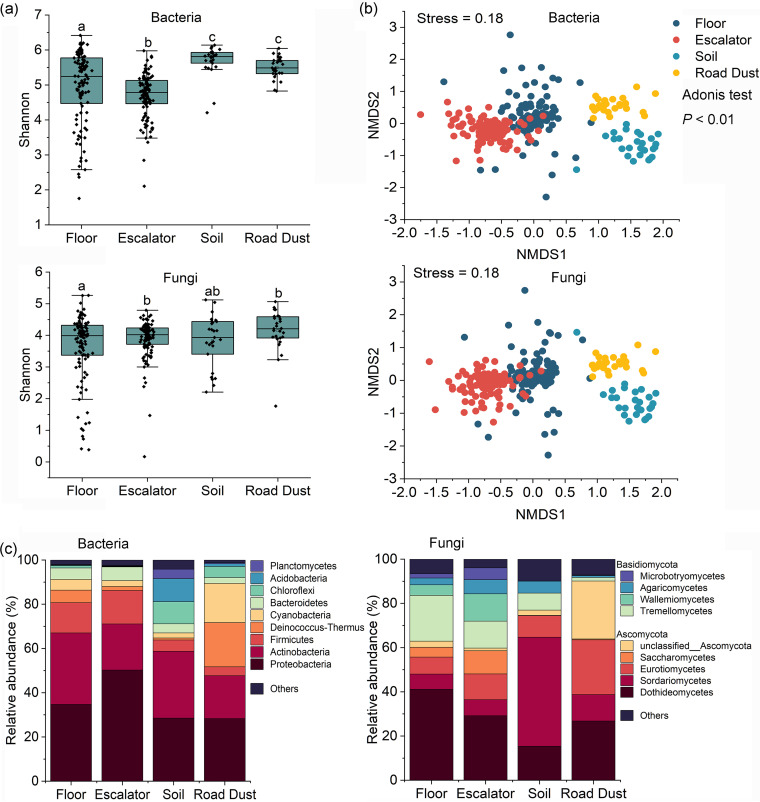
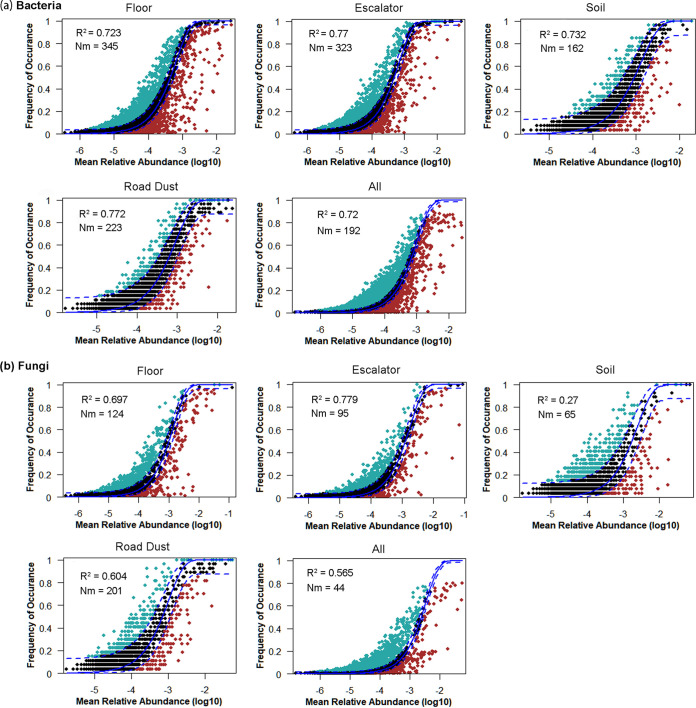
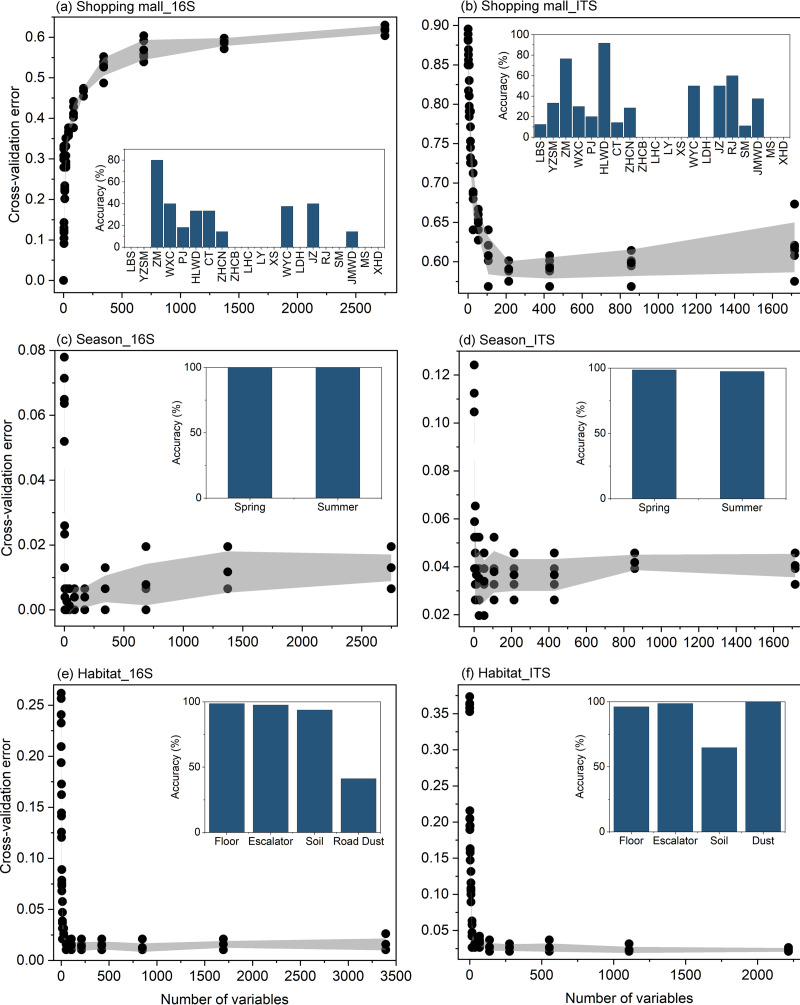
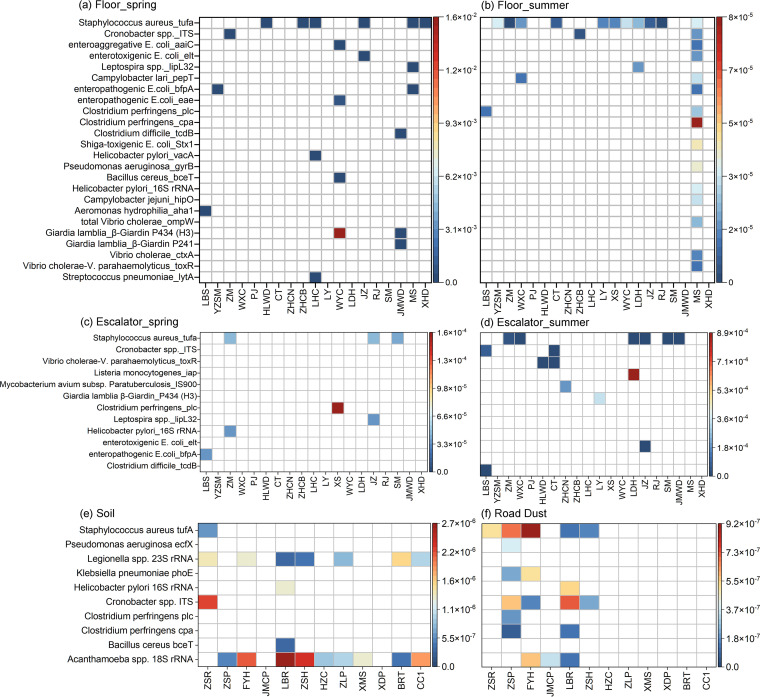
Shopping malls offer various niches for microbial populations, potentially serving as sources and reservoirs for the spread of microorganisms of public health concern. However, knowledge about the microbiome and the distribution of human pathogens in malls is largely unknown. Here, we examine the microbial community dynamics and genotypes of potential pathogens from floor and escalator surfaces in shopping malls and adjacent road dusts and greenbelt soils. The distribution pattern of microbial communities is driven primarily by habitats and seasons. A significant enrichment of human-associated microbiota in the indoor environment indicates that human interactions with surfaces might be another strong driver for mall microbiomes. Neutral community models suggest that the microbial community assembly is strongly driven by stochastic processes. Distinct performances of microbial taxonomic signatures for environmental classifications indicate the consistent differences of microbial communities of different seasons/habitats and the strong anthropogenic effect on homogenizing microbial communities of shopping malls. Indoor environments harbored higher concentrations of human pathogens than outdoor samples, also carrying a high proportion of antimicrobial resistance-associated multidrug efflux genes and virulence genes. These findings enhanced the understanding of the microbiome in the built environment and the interactions between humans and the built environment, providing a basis for tracking biothreats and communicable diseases and developing sophisticated early warning systems. IMPORTANCE Shopping malls are distinct microbial environments which can facilitate a constant transmission of microorganisms of public health concern between humans and the built environment or between human and human. Despite extensive investigation of the natural environmental microbiome, no comprehensive profile of microbial ecology has been reported in malls. Characterizing microbial distribution, potential pathogens, and antimicrobial resistance will enhance our understanding of how these microbial communities are formed, maintained, and transferred and help establish a baseline for biosurveillance of potential public health threats in malls.
Keywords: antimicrobial resistance; built environment; human pathogen; microbial assembly; microbial interactions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hsu T, Joice R, Vallarino J, Abu-Ali G, Hartmann EM, Shafquat A, DuLong C, Baranowski C, Gevers D, Green JL, Morgan XC, Spengler JD, Huttenhower C. 2016. Urban transit system microbial communities differ by surface type and interaction with humans and the environment. mSystems 1:e00018-16. doi: 10.1128/mSystems.00018-16. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
