

Targeting the DNA damage response for cancer therapy
- PMID: 36606678
- PMCID: PMC9988002
- DOI: 10.1042/BST20220681
Targeting the DNA damage response for cancer therapy
Abstract
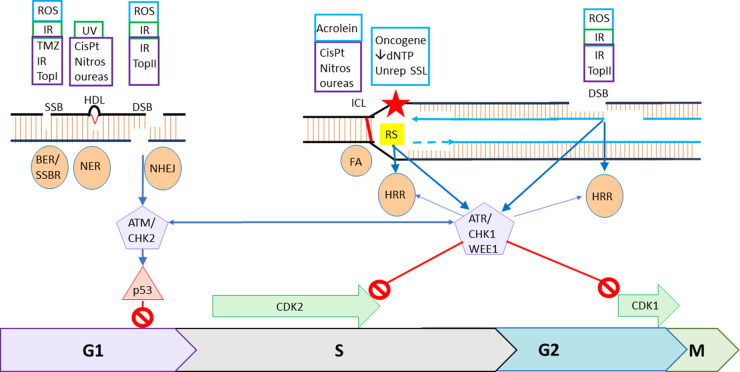
The DNA damage response (DDR) is an elegant system, coordinating DNA repair with cell cycle checkpoints, that evolved to protect living organisms from the otherwise fatal levels of DNA damage inflicted by endogenous and environmental sources. Since many agents used to treat cancer; radiotherapy and cytotoxic chemotherapy, work by damaging DNA the DDR represents a mechanism of resistance. The original rational for the development of drugs to inhibit the DDR was to overcome this mechanism of resistance but clinical studies using this approach have not led to improvements in the therapeutic index. A more exciting approach is to exploit cancer-specific defects in the DDR, that represent vulnerabilities in the tumour and an opportunity to selectively target the tumour. PARP inhibitors (PARPi) selectively kill homologous recombination repair defective (HRD, e.g. through BRCA mutation) cells. This approach has proven successful clinically and there are now six PARPi approved for cancer therapy. Drugs targeting other aspects of the DDR are under pre-clinical and clinical evaluation as monotherapy agents and in combination studies. For this promising approach to cancer therapy to be fully realised reliable biomarkers are needed to identify tumours with the exploitable defect for monotherapy applications. The possibility that some combinations may result in toxicity to normal tissues also needs to be considered. A brief overview of the DDR, the development of inhibitors targeting the DDR and the current clinical status of such drugs is described here.
Keywords: ATM; ATR; CHK1; DNA damage response; DNA-PK; PARP.
© 2023 The Author(s).
Conflict of interest statement
The author was involved in the development of rucaparib and has received royalty payments but does not take them personally. She has also been involved in the development of DNA-PK inhibitors (NU7XXX series) and ATM inhibitors (KU55933 etc) and also diverts any royalty payments elsewhere. She identified NU6027 as an ATR inhibitor but this has never been commercialised.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous