

Hereditary Angioedema: Diagnosis, Clinical Implications, and Pathophysiology
- PMID: 36609679
- PMCID: PMC9988798
- DOI: 10.1007/s12325-022-02401-0
Hereditary Angioedema: Diagnosis, Clinical Implications, and Pathophysiology
Abstract
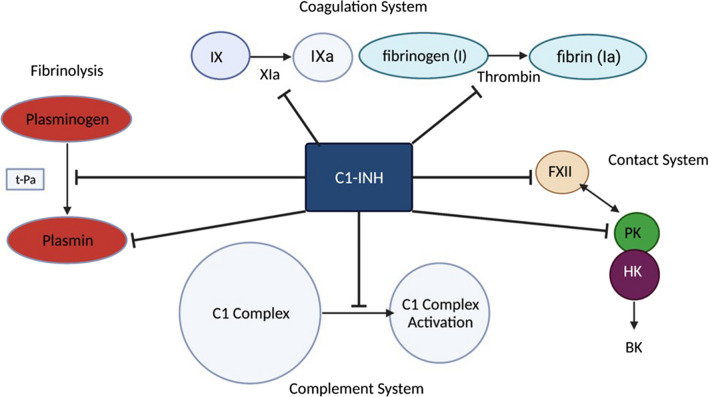
Hereditary angioedema (HAE) is an autosomal dominant disorder caused by a mutation in the C1 esterase inhibitor gene. HAE affects 1/50,000 people worldwide. Three main types of HAE exist: type I, type II, and type III. Type I is characterized by a deficiency in C1-INH. C1-INH is important in the coagulation complement, contact systems, and fibrinolysis. Most HAE cases are type I. Type I and II HAE result from a mutation in the SERPING1 gene, which encodes C1-INH. Formally known as type III HAE is typically an estrogen-dependent or hereditary angioedema with normal C1-INH activity. Current guidelines now recommend subdividing hereditary angioedema with normal C1 esterase inhibitor gene (HAE-nl-C1-INH formerly known as HAE type III) based on underlying mutations such as in kininogen-1 (HAE-KNG1), plasminogen gene (PLG-HAE), myoferlin gene mutation (MYOF-HAE), heparan sulfate-glucosamine 3-sulfotransferase 6 (HS3ST6), mutation in Hageman factor (factor XII), and in angiopoietin-1 (HAE-ANGPT-1). The clinical presentation of HAE varies between patients, but it usually presents with nonpitting angioedema and occasionally abdominal pain. Young children are typically asymptomatic. Those affected by HAE usually present with symptoms in their early 20s. Symptoms can arise as a result of stress, infection, or trauma. Laboratory testing shows abnormal levels of C1-INH and high levels of bradykinin. C4 and D-dimer levels can also be monitored if an acute HAE attack is suspected. Acute treatment of HAE can include IV infusions of C1-INH, receptor antagonists, and kallikrein inhibitors. Short- and long-term prophylaxis can also be administered to patients with HAE. First-line therapies for long-term prophylaxis also include IV infusion of C1-INH. This review aims to thoroughly understand HAE, its clinical presentation, and how to treat it.
Keywords: Angioedema; C1-INH; F12; Genetics; HAE; Hereditary; SERPING1.
© 2023. The Author(s).
Figures
Similar articles
-
The Genetics of Hereditary Angioedema: A Review.J Clin Med. 2021 May 9;10(9):2023. doi: 10.3390/jcm10092023. J Clin Med. 2021. PMID: 34065094 Free PMC article. Review.
-
The Expanding Spectrum of Mutations in Hereditary Angioedema.J Allergy Clin Immunol Pract. 2021 Jun;9(6):2229-2234. doi: 10.1016/j.jaip.2021.03.008. Epub 2021 Mar 19. J Allergy Clin Immunol Pract. 2021. PMID: 33746090
-
Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence.Orphanet J Rare Dis. 2020 Oct 15;15(1):289. doi: 10.1186/s13023-020-01570-x. Orphanet J Rare Dis. 2020. PMID: 33059692 Free PMC article.
-
Diagnosis and treatment of hereditary angioedema.Panminerva Med. 2012 Sep;54(3):241-53. Panminerva Med. 2012. PMID: 22801442 Review.
-
Screening for Plasminogen Mutations in Hereditary Angioedema Patients.Genes (Basel). 2021 Mar 11;12(3):402. doi: 10.3390/genes12030402. Genes (Basel). 2021. PMID: 33799813 Free PMC article.
Cited by
-
COVID-19 and vaccination in hereditary angioedema: Single center experience.World Allergy Organ J. 2024 Mar 23;17(4):100892. doi: 10.1016/j.waojou.2024.100892. eCollection 2024 Apr. World Allergy Organ J. 2024. PMID: 38559494 Free PMC article.
-
Identification of an elusive SERPING1 deletion in a family with hereditary angioedema type I utilizing soft clipping.Front Allergy. 2025 Apr 17;6:1565283. doi: 10.3389/falgy.2025.1565283. eCollection 2025. Front Allergy. 2025. PMID: 40313637 Free PMC article.
-
Case Report: Early presentation of hereditary angioedema symptoms in a 2-year-old boy.Front Pediatr. 2024 Jun 24;12:1408110. doi: 10.3389/fped.2024.1408110. eCollection 2024. Front Pediatr. 2024. PMID: 38978843 Free PMC article.
-
Analysis of prodromal symptoms and need for short-term prophylaxis in angioedema patients under long-term prophylaxis.Orphanet J Rare Dis. 2025 Feb 1;20(1):47. doi: 10.1186/s13023-025-03562-1. Orphanet J Rare Dis. 2025. PMID: 39893484 Free PMC article.
-
Cadmium Exposure Disrupts Uterine Energy Metabolism and Coagulation Homeostasis During Labor in Institute of Cancer Research Mice: Insights from Transcriptomic Analysis.Metabolites. 2025 May 20;15(5):339. doi: 10.3390/metabo15050339. Metabolites. 2025. PMID: 40422915 Free PMC article.
References
-
- Andrejević S, Korošec P, Šilar M, Košnik M, Mijanović R, Bonači-Nikolić B, et al. Hereditary angioedema due to C1 inhibitor deficiency in Serbia: two novel mutations and evidence of genotype–phenotype association. PLoS ONE. 2015;10(11):e0142174. doi: 10.1371/journal.pone.0142174. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
