

Fast, accurate local ancestry inference with FLARE
- PMID: 36610402
- PMCID: PMC9943733
- DOI: 10.1016/j.ajhg.2022.12.010
Fast, accurate local ancestry inference with FLARE
Abstract
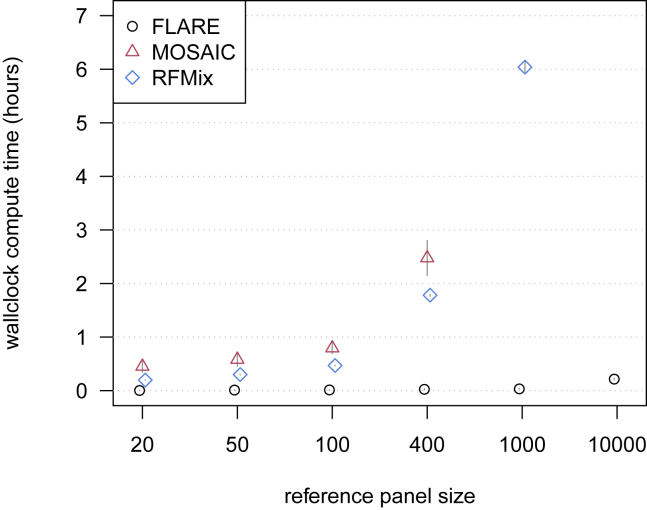
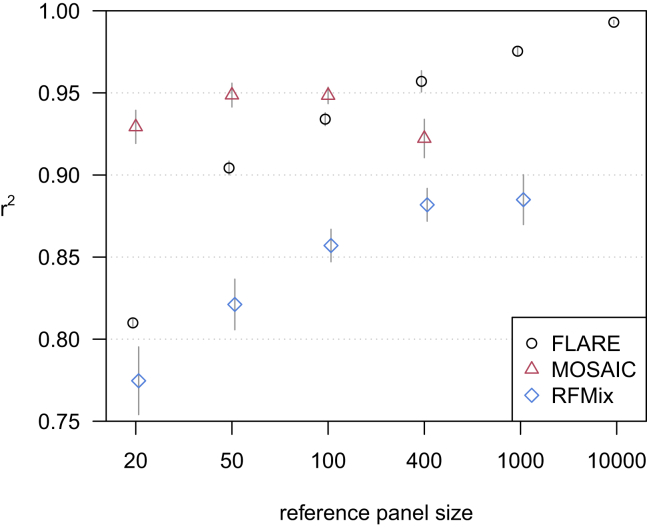
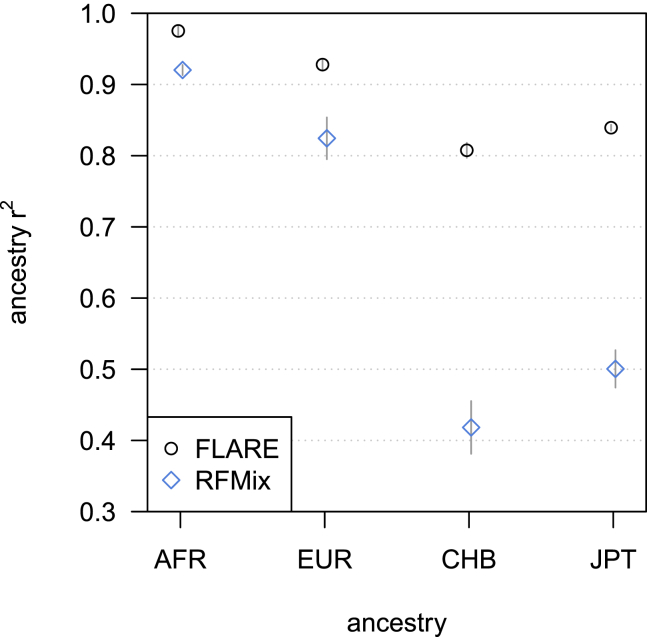
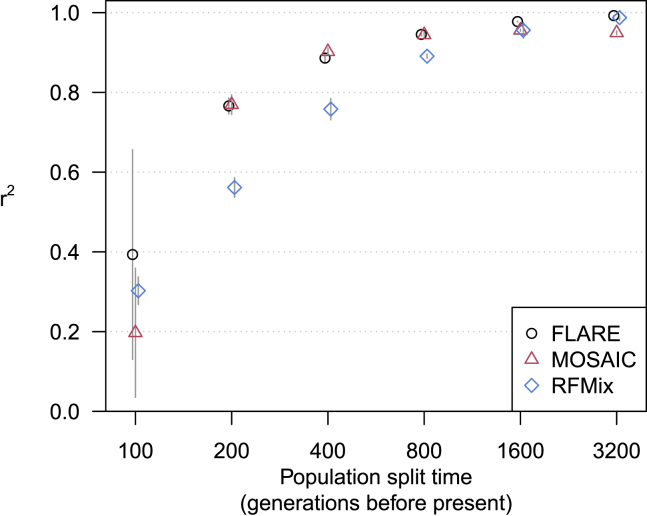
Local ancestry is the source ancestry at each point in the genome of an admixed individual. Inferred local ancestry is used for admixture mapping and population genetic analyses. We present FLARE (fast local ancestry estimation), a method for local ancestry inference. FLARE achieves high accuracy through the use of an extended Li and Stephens model, and it achieves exceptional computational performance through incorporation of computational techniques developed for genotype imputation. Memory requirements are reduced through on-the-fly compression of reference haplotypes and stored checkpoints. Computation time is reduced through the use of composite reference haplotypes. These techniques allow FLARE to scale to datasets with hundreds of thousands of sequenced individuals and to provide superior accuracy on large-scale data. FLARE is open source and available at https://github.com/browning-lab/flare.
Keywords: admixture; ancestry; local ancestry inference.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
