

The Role of Proteolysis in Amyloidosis
- PMID: 36614141
- PMCID: PMC9820691
- DOI: 10.3390/ijms24010699
The Role of Proteolysis in Amyloidosis
Abstract
Amyloidoses are a group of diseases associated with deposits of amyloid fibrils in different tissues. So far, 36 different types of amyloidosis are known, each due to the misfolding and accumulation of a specific protein. Amyloid deposits can be found in several organs, including the heart, brain, kidneys, and spleen, and can affect single or multiple organs. Generally, amyloid-forming proteins become prone to aggregate due to genetic mutations, acquired environmental factors, excessive concentration, or post-translational modifications. Interestingly, amyloid aggregates are often composed of proteolytic fragments, derived from the degradation of precursor proteins by yet unidentified proteases, which display higher amyloidogenic tendency compared to precursor proteins, thus representing an important mechanism in the onset of amyloid-based diseases. In the present review, we summarize the current knowledge on the proteolytic susceptibility of three of the main human amyloidogenic proteins, i.e., transthyretin, β-amyloid precursor protein, and α-synuclein, in the onset of amyloidosis. We also highlight the role that proteolytic enzymes can play in the crosstalk between intestinal inflammation and amyloid-based diseases.
Keywords: amyloid precursor protein; amyloidosis; protein aggregation; proteolysis; synuclein; transthyretin.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
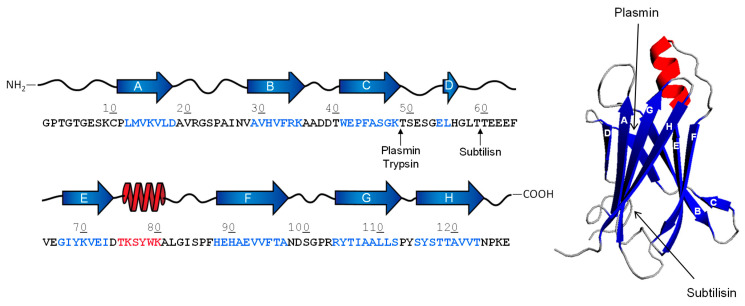
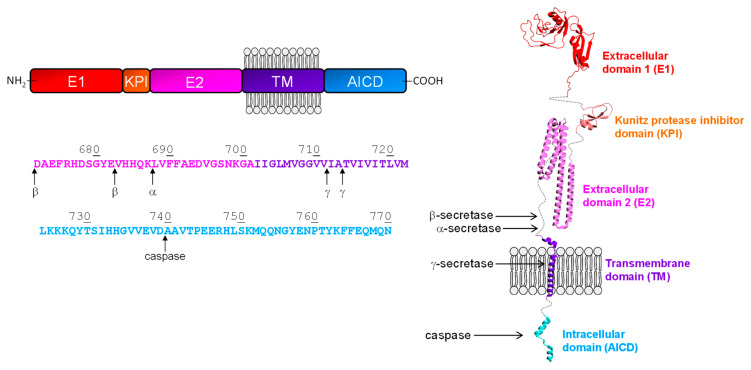
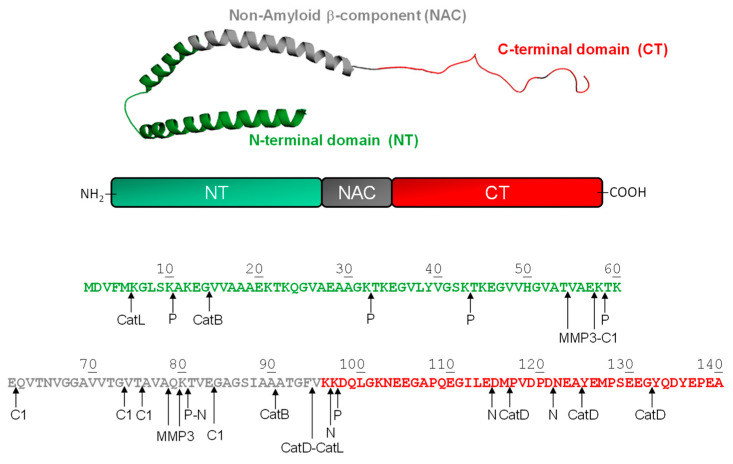
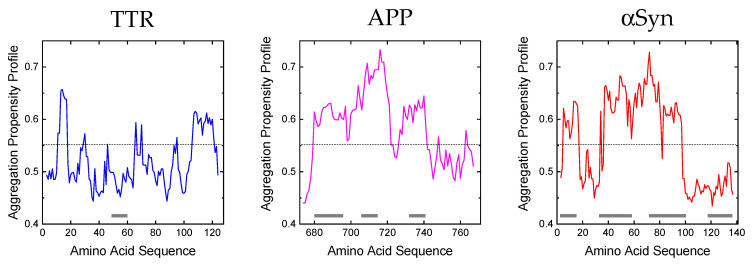
References
-
- Benson M.D., Buxbaum J.N., Eisenberg D.S., Merlini G., Saraiva M.J.M., Sekijima Y., Sipe J.D., Westermark P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2020;27:217–222. doi: 10.1080/13506129.2020.1835263. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
