

Traditional Machine and Deep Learning for Predicting Toxicity Endpoints
- PMID: 36615411
- PMCID: PMC9822478
- DOI: 10.3390/molecules28010217
Traditional Machine and Deep Learning for Predicting Toxicity Endpoints
Abstract
Molecular structure property modeling is an increasingly important tool for predicting compounds with desired properties due to the expensive and resource-intensive nature and the problem of toxicity-related attrition in late phases during drug discovery and development. Lately, the interest for applying deep learning techniques has increased considerably. This investigation compares the traditional physico-chemical descriptor and machine learning-based approaches through autoencoder generated descriptors to two different descriptor-free, Simplified Molecular Input Line Entry System (SMILES) based, deep learning architectures of Bidirectional Encoder Representations from Transformers (BERT) type using the Mondrian aggregated conformal prediction method as overarching framework. The results show for the binary CATMoS non-toxic and very-toxic datasets that for the former, almost equally balanced, dataset all methods perform equally well while for the latter dataset, with an 11-fold difference between the two classes, the MolBERT model based on a large pre-trained network performs somewhat better compared to the rest with high efficiency for both classes (0.93-0.94) as well as high values for sensitivity, specificity and balanced accuracy (0.86-0.87). The descriptor-free, SMILES-based, deep learning BERT architectures seem capable of producing well-balanced predictive models with defined applicability domains. This work also demonstrates that the class imbalance problem is gracefully handled through the use of Mondrian conformal prediction without the use of over- and/or under-sampling, weighting of classes or cost-sensitive methods.
Keywords: BERT; CATMoS dataset; CDDD; RDKit; conformal prediction; random forest.
Conflict of interest statement
The author declares no conflict of interest.
Figures
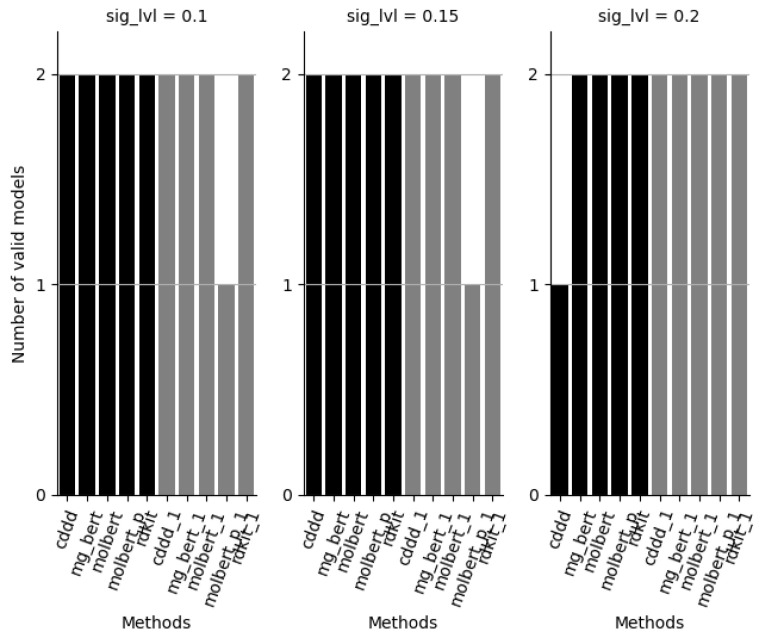
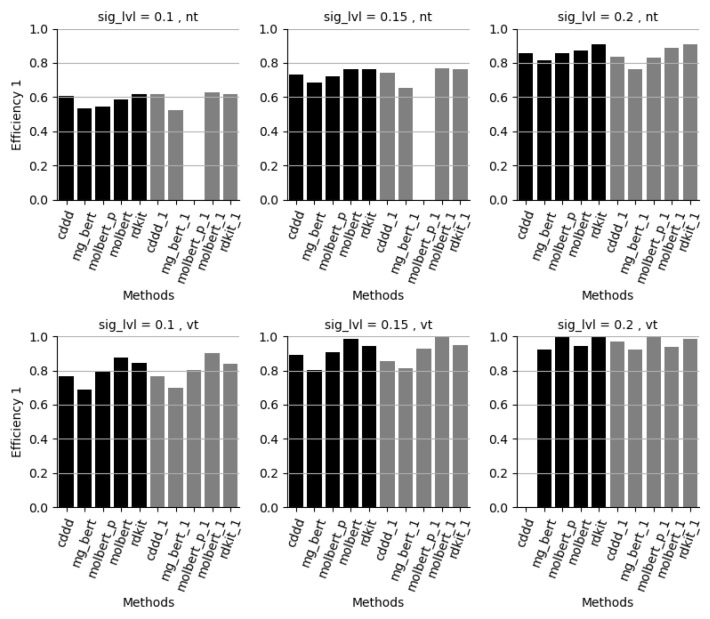
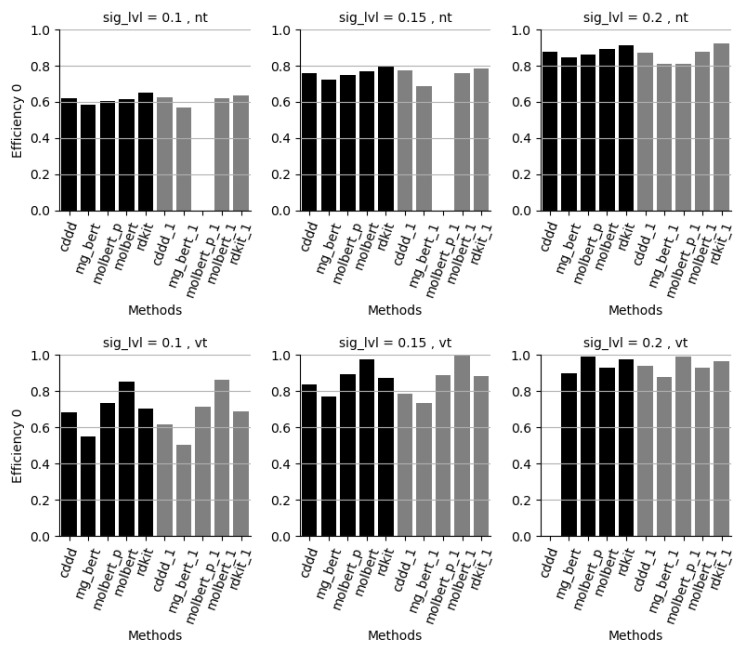
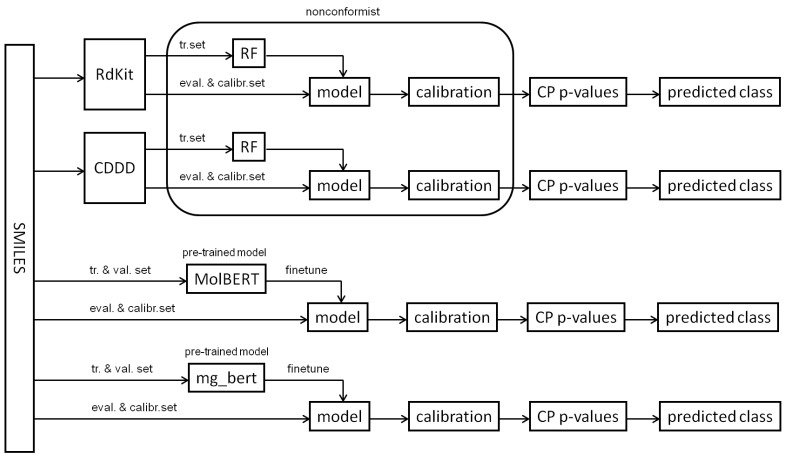
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
