

Balancing selection on genomic deletion polymorphisms in humans
- PMID: 36625544
- PMCID: PMC9943071
- DOI: 10.7554/eLife.79111
Balancing selection on genomic deletion polymorphisms in humans
Abstract
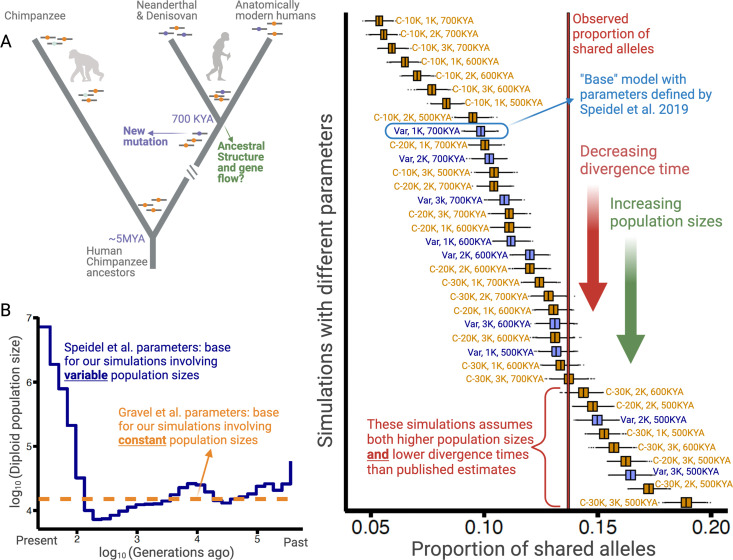
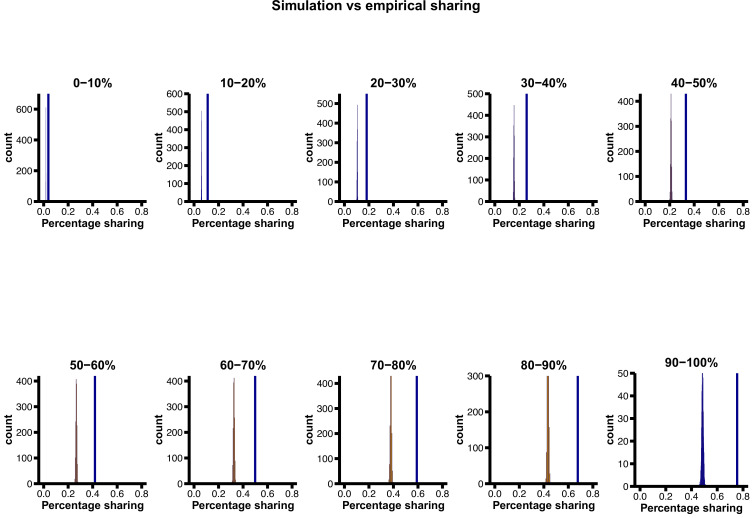
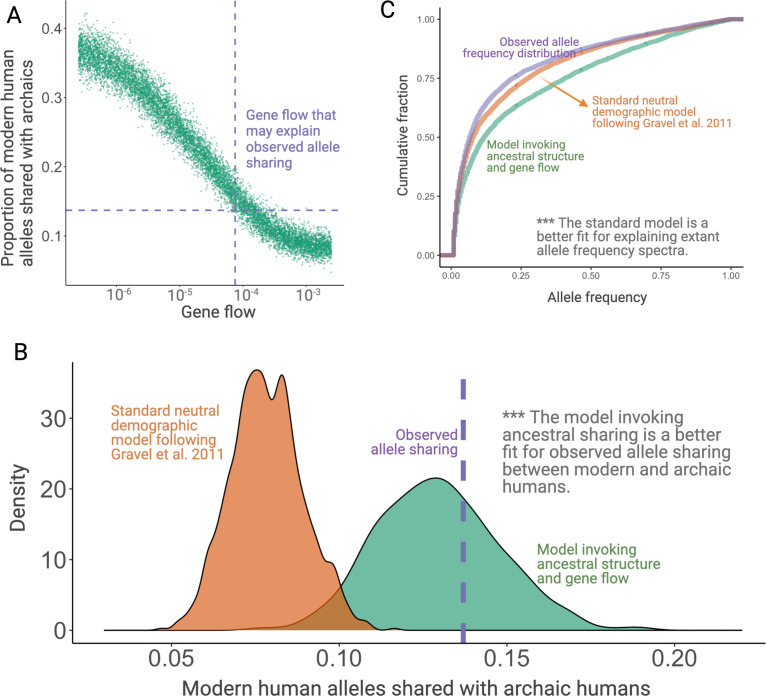
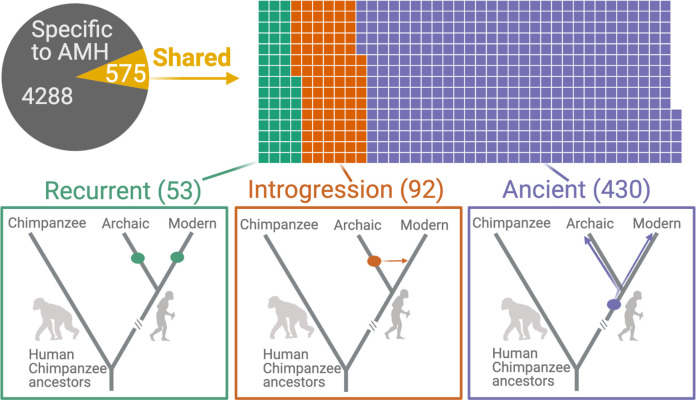
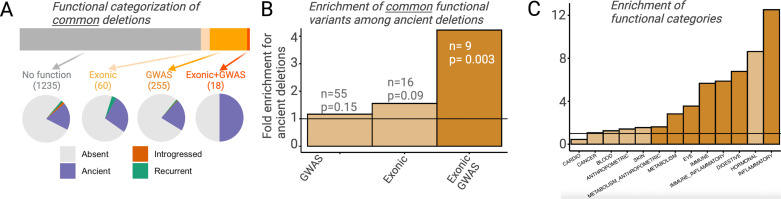
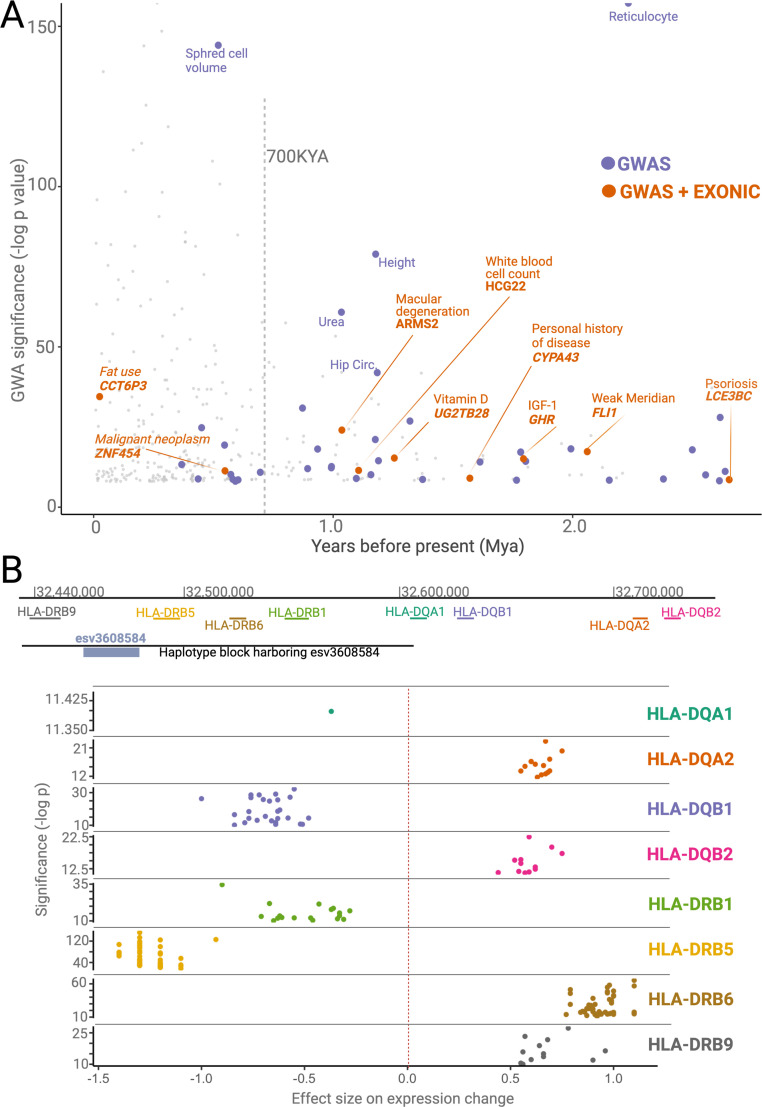
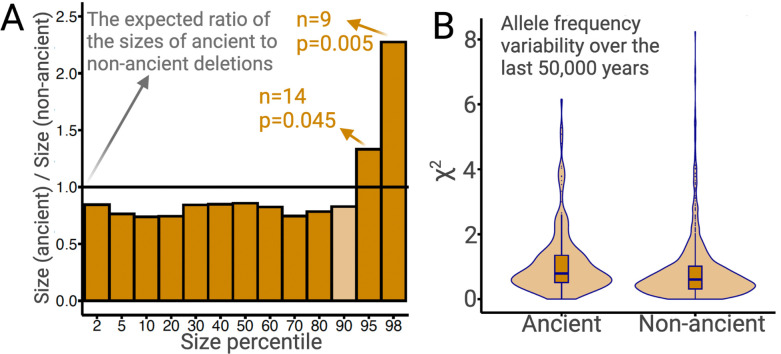
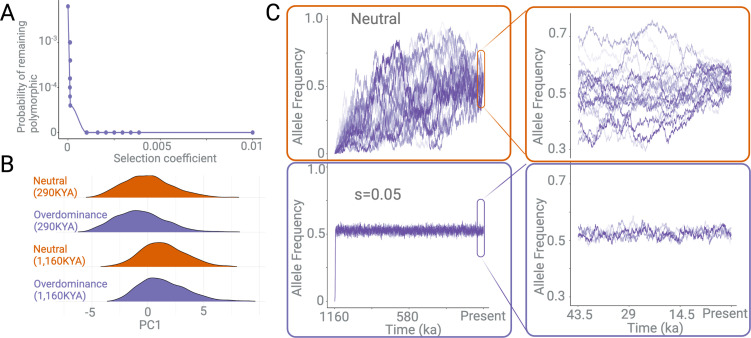
A key question in biology is why genomic variation persists in a population for extended periods. Recent studies have identified examples of genomic deletions that have remained polymorphic in the human lineage for hundreds of millennia, ostensibly owing to balancing selection. Nevertheless, genome-wide investigation of ancient and possibly adaptive deletions remains an imperative exercise. Here, we demonstrate an excess of polymorphisms in present-day humans that predate the modern human-Neanderthal split (ancient polymorphisms), which cannot be explained solely by selectively neutral scenarios. We analyze the adaptive mechanisms that underlie this excess in deletion polymorphisms. Using a previously published measure of balancing selection, we show that this excess of ancient deletions is largely owing to balancing selection. Based on the absence of signatures of overdominance, we conclude that it is a rare mode of balancing selection among ancient deletions. Instead, more complex scenarios involving spatially and temporally variable selective pressures are likely more common mechanisms. Our results suggest that balancing selection resulted in ancient deletions harboring disproportionately more exonic variants with GWAS (genome-wide association studies) associations. We further found that ancient deletions are significantly enriched for traits related to metabolism and immunity. As a by-product of our analysis, we show that deletions are, on average, more deleterious than single nucleotide variants. We can now argue that not only is a vast majority of common variants shared among human populations, but a considerable portion of biologically relevant variants has been segregating among our ancestors for hundreds of thousands, if not millions, of years.
Keywords: Denisovans; Neanderthals; copy number variation; evolution; evolutionary biology; genetics; genomics; human; structural variation.
Plain language summary
The persistence of versions of genes that cause severe disease in human populations has long perplexed scientists. It is common for many versions of a gene to exist. But scientists expect that over time natural selection will eliminate versions of genes harmful to human health. Sometimes, there are good reasons that a disease-causing gene may persist. For example, having two copies of a particular gene variant causes a condition, called sickle cell disease. But having one sickle cell-causing copy of the gene and one non-disease-causing copy protects against malaria. As a result, the version of the gene that causes sickle cell is more common in people from areas where malaria is prevalent despite the risks to people who end up with two copies. Scientists call this phenomenon balancing selection because trade-offs in the gene’s benefits and risks cause it to persist in the population. Aqil et al. show that balancing selection has likely caused many ancient gene variants to persist in human populations. In the experiments, Aqil et al. scoured the genomes of hundreds of modern humans from around the world and four groups of ancient human ancestors, including Neanderthals and Denisovans. The experiments looked for structural changes in genes, like deletions, that date back to more than 700,000 years ago – before modern humans split from their ancestors. They found large numbers of such ancient genes in modern humans. Using computer modeling, Aqil et al. showed that these ancient genes likely persist because of balancing selection. Many of these ancient genes regulate the immune response and metabolism. These genes may protect against infectious diseases outbreaks and starvation, which have occurred periodically throughout human history. But these same genes may cause immune or metabolic diseases in modern humans not currently facing these threats. The experiments show how such biological trade-offs have shaped human evolution and reveal that modern human populations, regardless of race or region of origin, share the same genetic variation that already our ancestors carried within them.
© 2023, Aqil et al.
Conflict of interest statement
AA, LS, PP, OG No competing interests declared
Figures
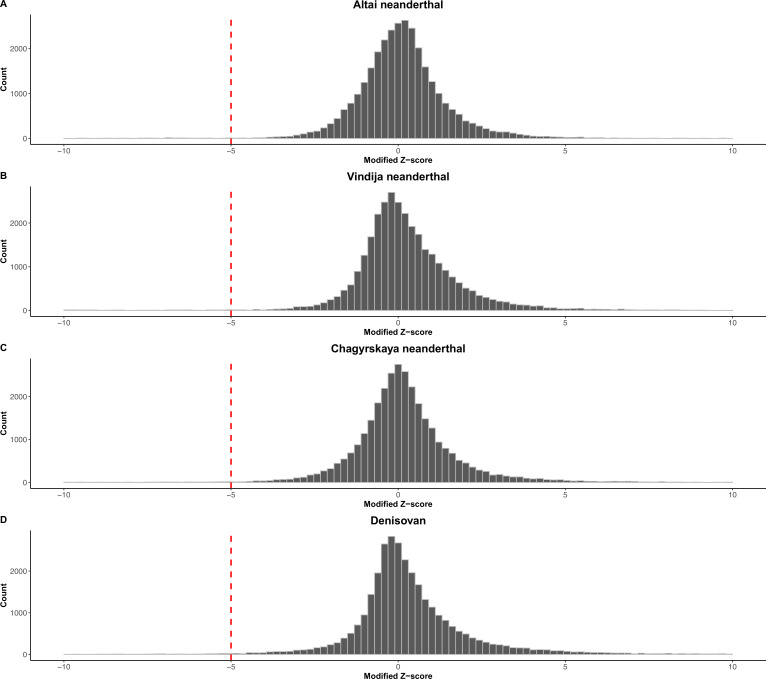
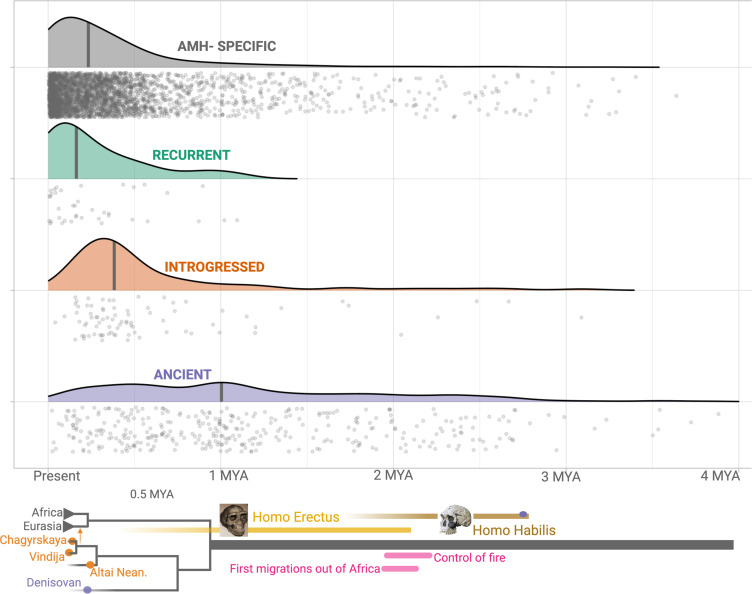
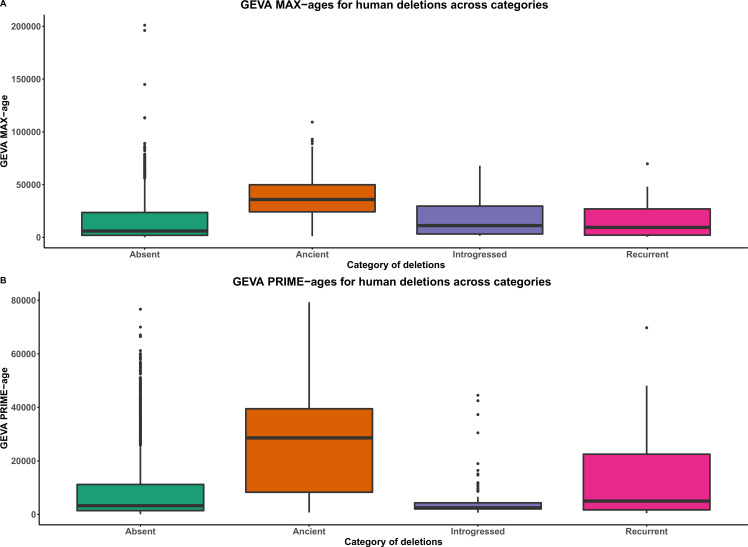
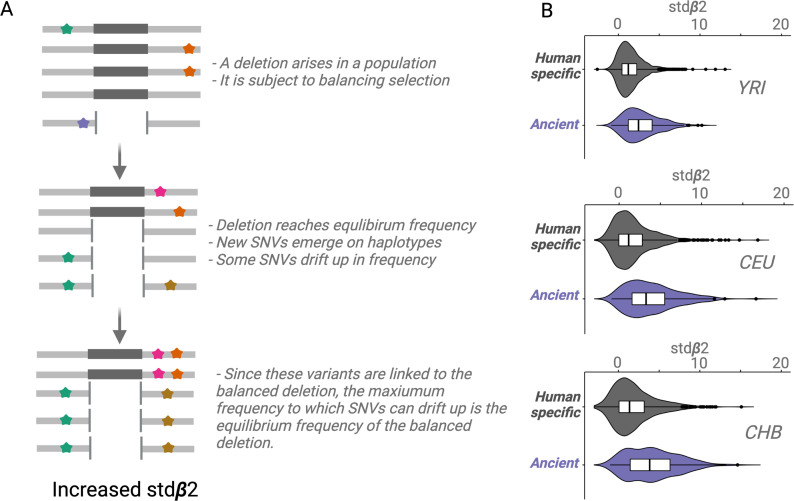
References
-
- Agarwal V, Kommaddi RP, Valli K, Ryder D, Hyde TM, Kleinman JE, Strobel HW, Ravindranath V. Drug metabolism in human brain: high levels of cytochrome P4503A43 in brain and metabolism of anti-anxiety drug alprazolam to its active metabolite. PLOS ONE. 2008;3:e2337. doi: 10.1371/journal.pone.0002337. - DOI - PMC - PubMed
-
- Aho AV, Kernighan BW, Weinberger PJ. Awk — a pattern scanning and processing language. Software. 1979;9:267–279. doi: 10.1002/spe.4380090403. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
