

Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis
- PMID: 36629024
- PMCID: PMC9832433
- DOI: 10.1002/ctm2.1170
Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis
Abstract
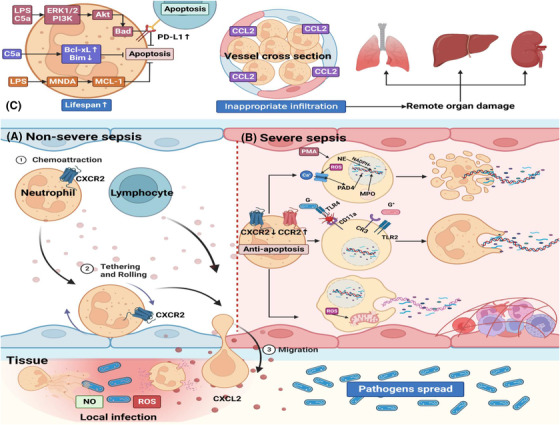
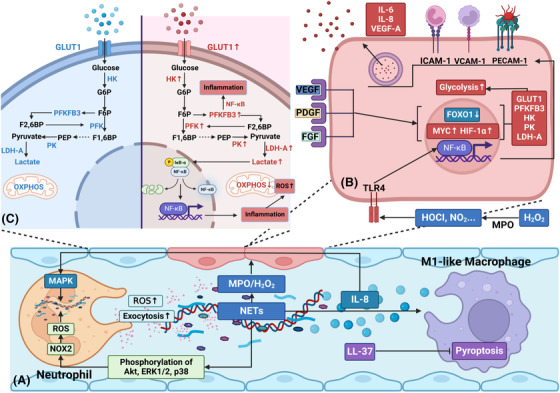
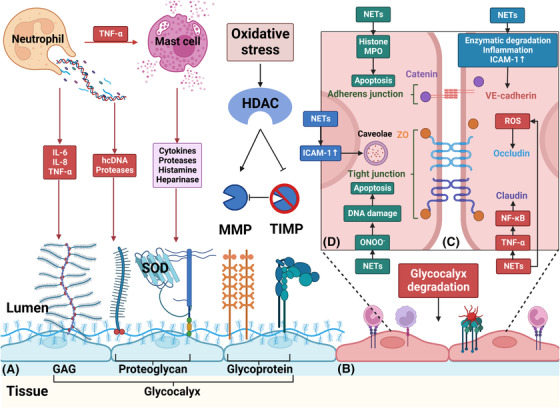
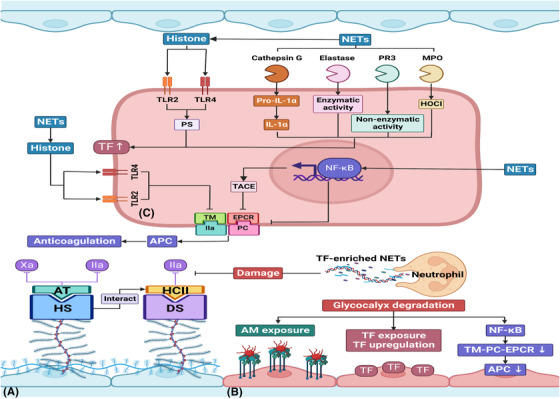
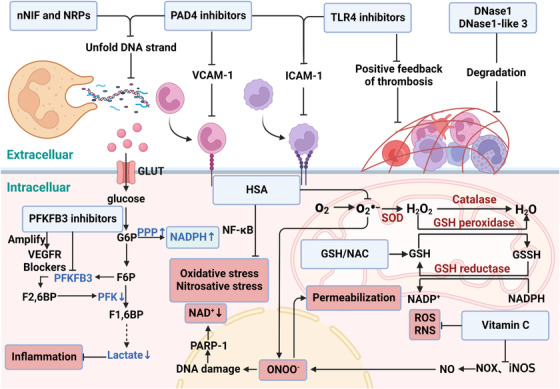
Sepsis is a persistent systemic inflammatory condition involving multiple organ failures resulting from a dysregulated immune response to infection, and one of the hallmarks of sepsis is endothelial dysfunction. During its progression, neutrophils are the first line of innate immune defence against infection. Aside from traditional mechanisms, such as phagocytosis or the release of inflammatory cytokines, reactive oxygen species and other antibacterial substances, activated neutrophils also release web-like structures composed of tangled decondensed DNA, histone, myeloperoxidase and other granules called neutrophil extracellular traps (NETs), which can efficiently ensnare bacteria in the circulation. In contrast, excessive neutrophil activation and NET release may induce endothelial cells to shift toward a pro-inflammatory and pro-coagulant phenotype. Furthermore, neutrophils and NETs can degrade glycocalyx on the endothelial cell surface and increase endothelium permeability. Consequently, the endothelial barrier collapses, contributing to impaired microcirculatory blood flow, tissue hypoperfusion and life-threatening organ failure in the late phase of sepsis.
Keywords: endothelial cell dysfunction; neutrophil; neutrophil extracellular traps; sepsis.
© 2023 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare that they have no conflict of interest
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials