

Uncovering perturbations in human hematopoiesis associated with healthy aging and myeloid malignancies at single-cell resolution
- PMID: 36629404
- PMCID: PMC9904760
- DOI: 10.7554/eLife.79363
Uncovering perturbations in human hematopoiesis associated with healthy aging and myeloid malignancies at single-cell resolution
Abstract
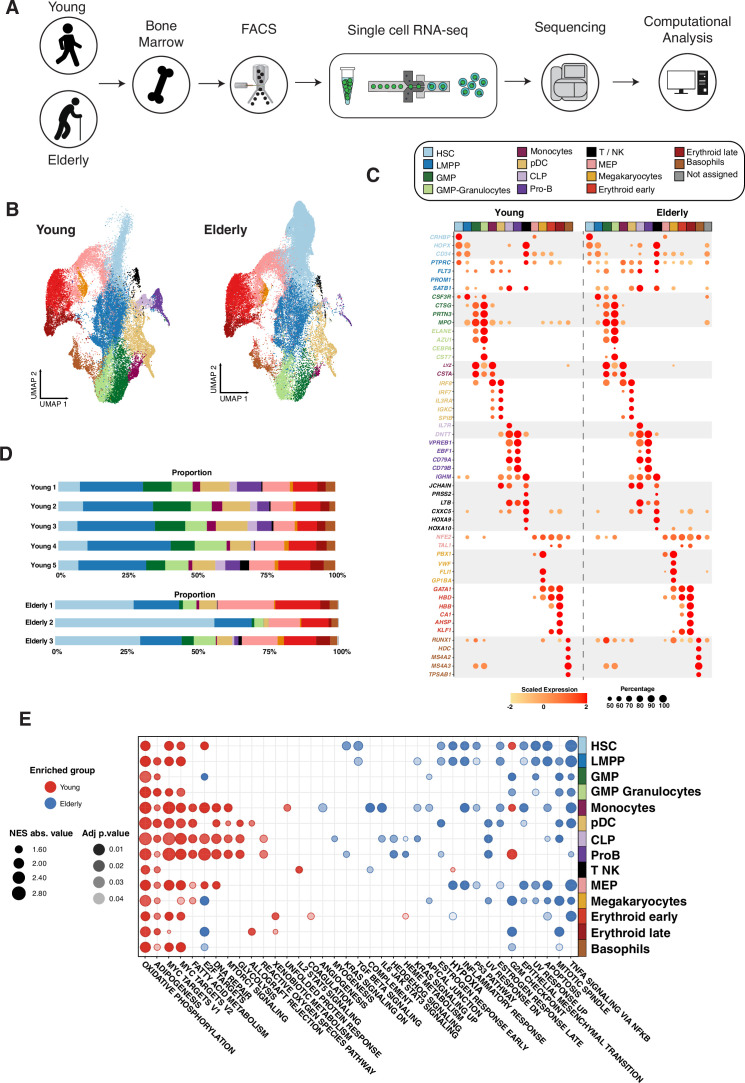
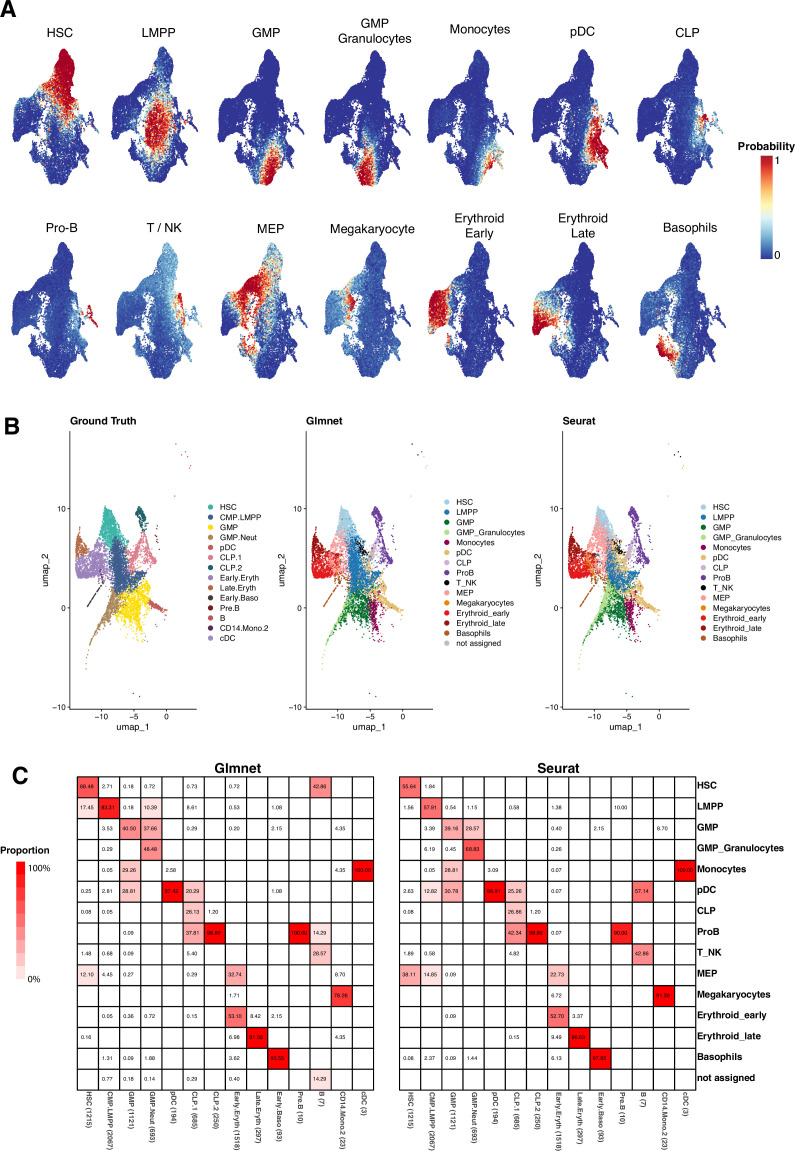
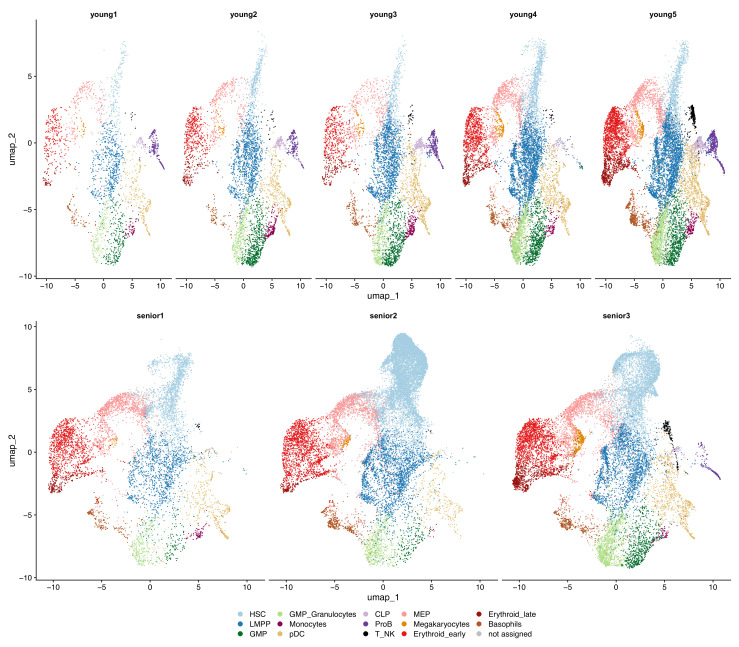
Early hematopoiesis is a continuous process in which hematopoietic stem and progenitor cells (HSPCs) gradually differentiate toward specific lineages. Aging and myeloid malignant transformation are characterized by changes in the composition and regulation of HSPCs. In this study, we used single-cell RNA sequencing (scRNA-seq) to characterize an enriched population of human HSPCs obtained from young and elderly healthy individuals.
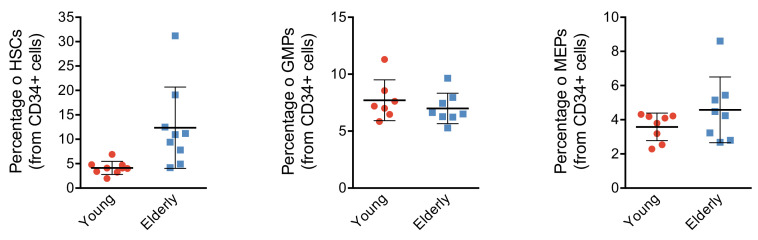
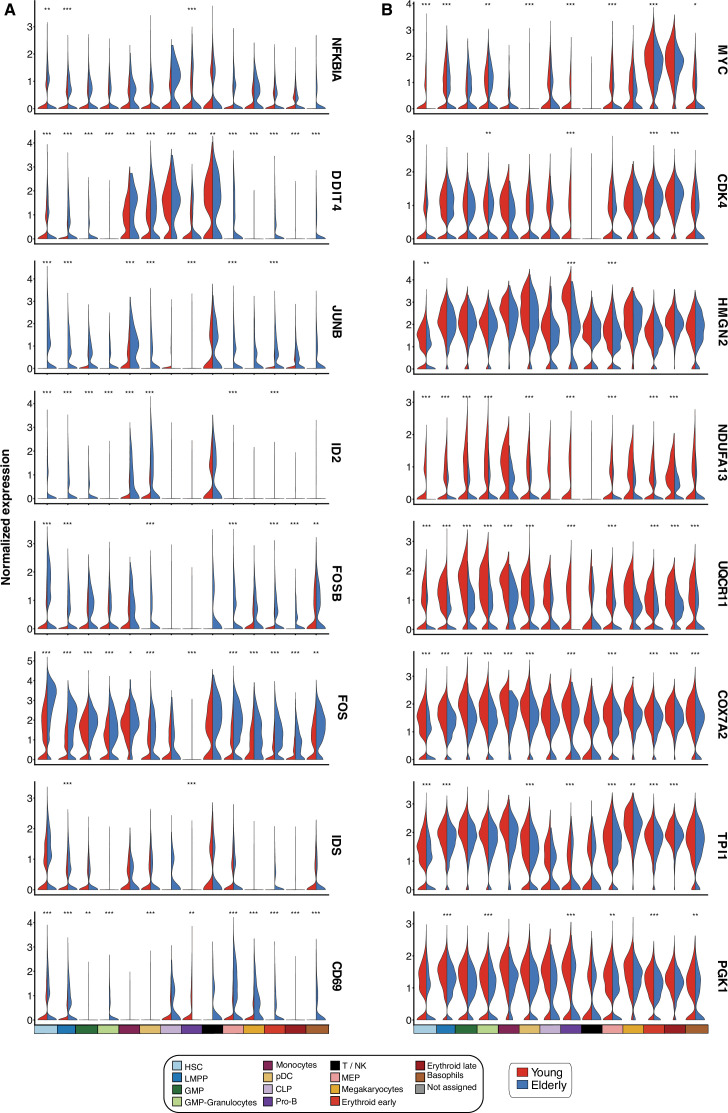
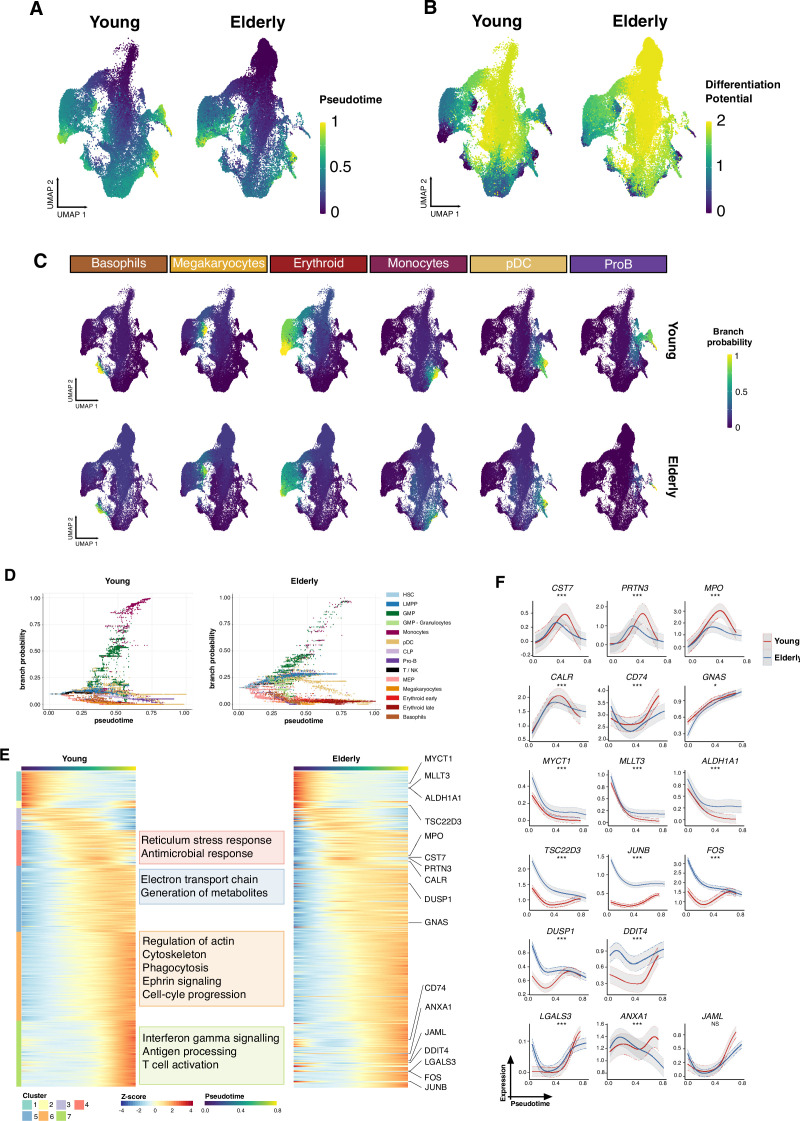
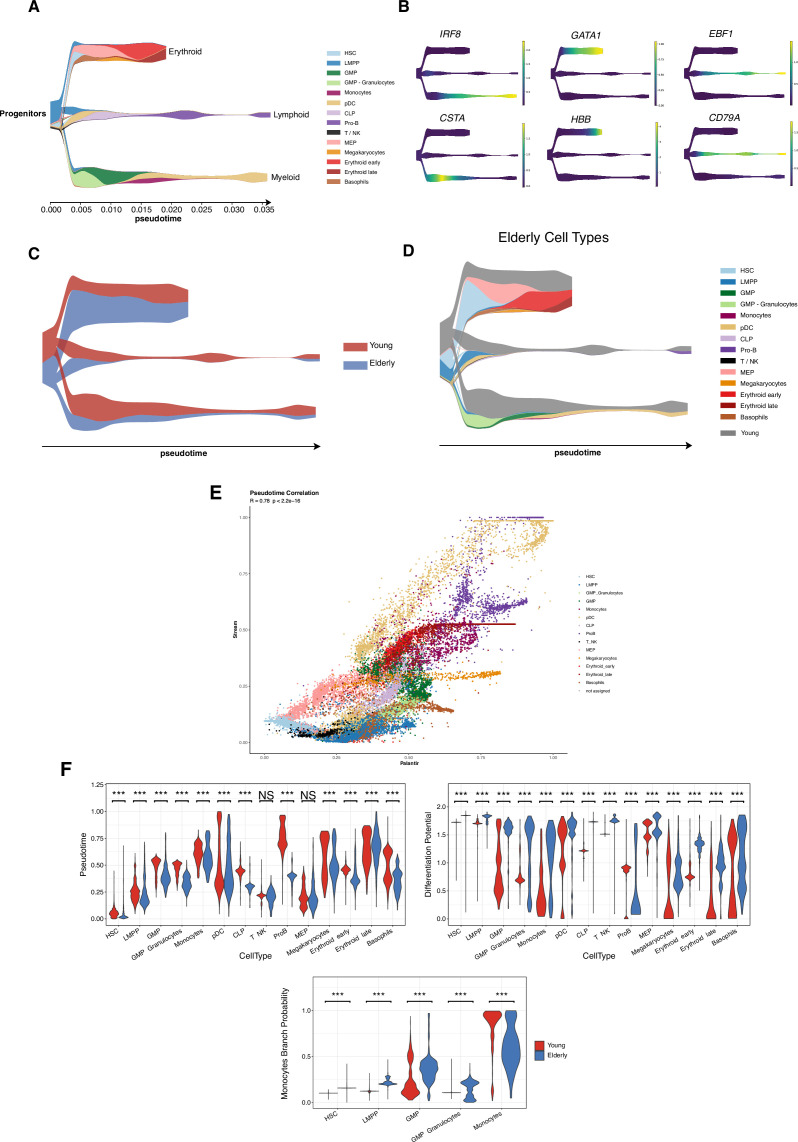
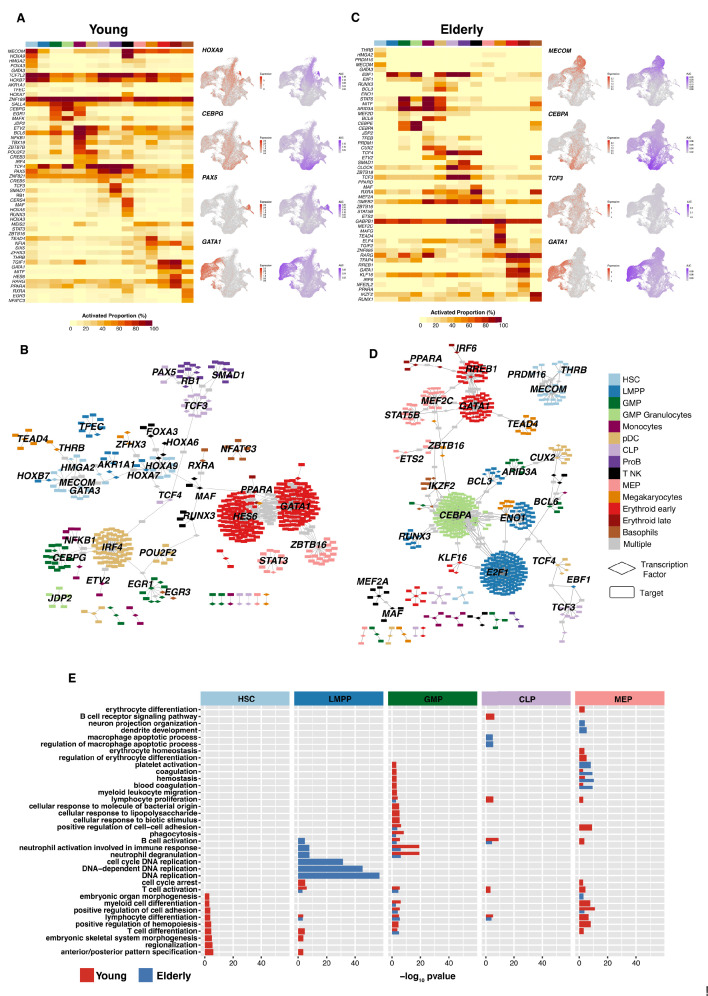
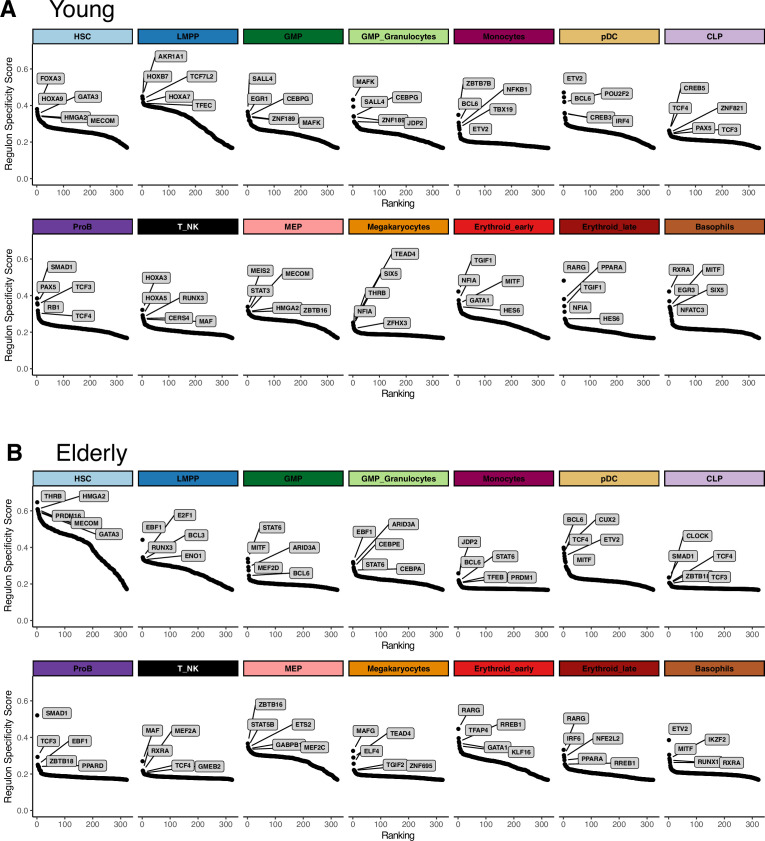
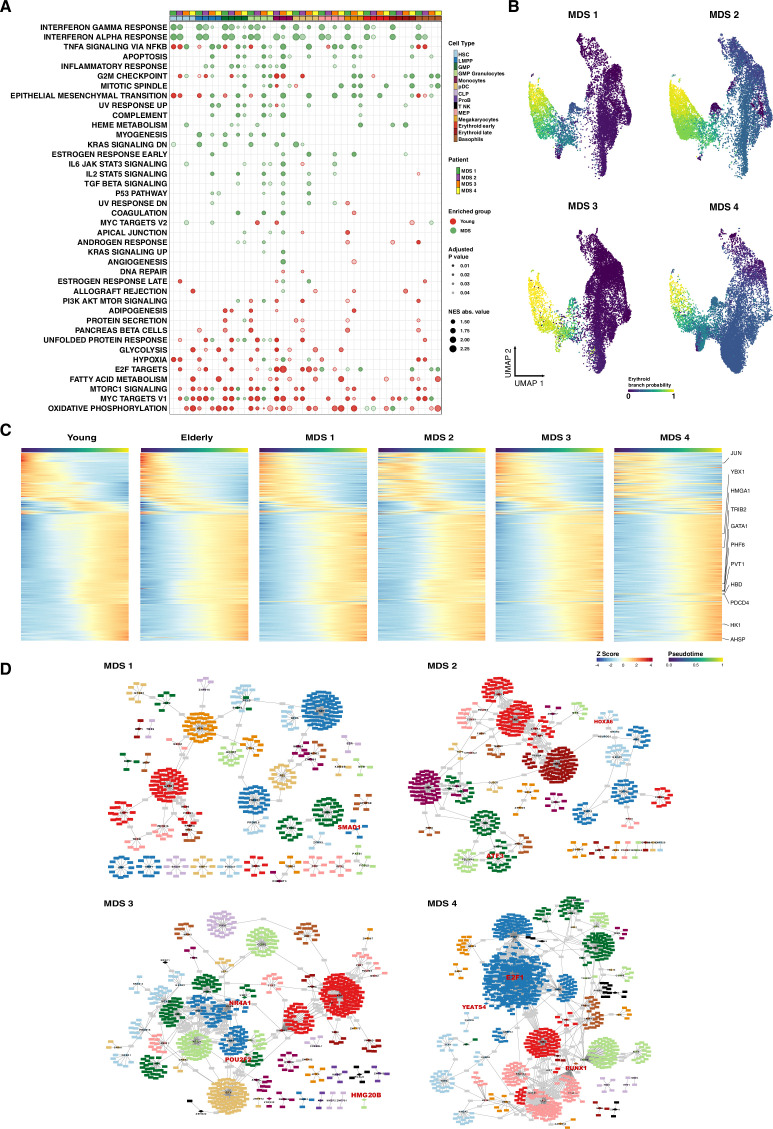
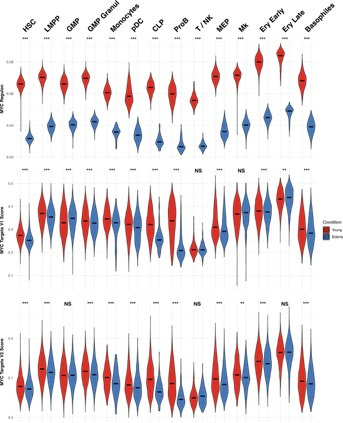
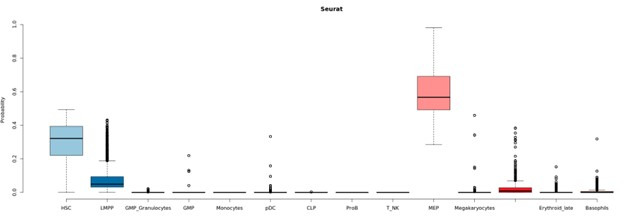
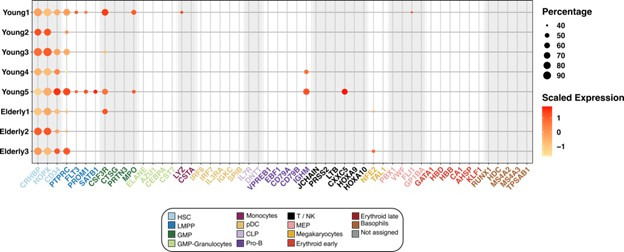
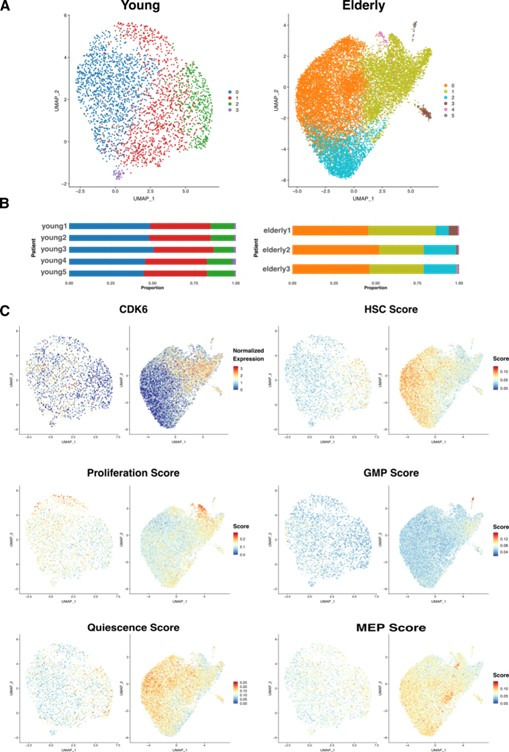
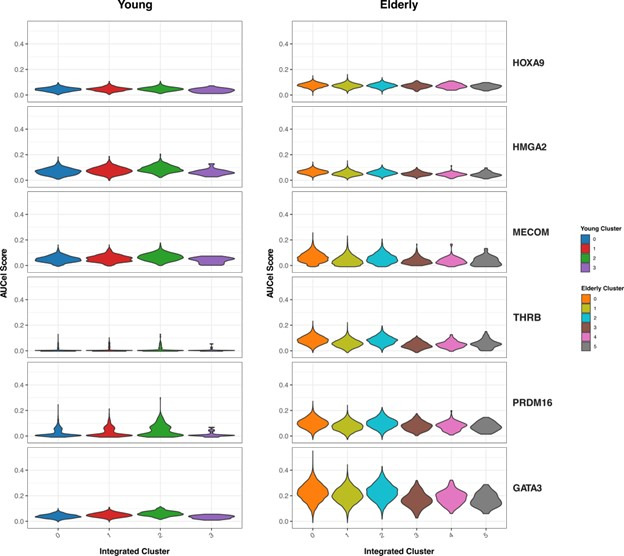
Based on their transcriptional profile, we identified changes in the proportions of progenitor compartments during aging, and differences in their functionality, as evidenced by gene set enrichment analysis. Trajectory inference revealed that altered gene expression dynamics accompanied cell differentiation, which could explain aging-associated changes in hematopoiesis. Next, we focused on key regulators of transcription by constructing gene regulatory networks (GRNs) and detected regulons that were specifically active in elderly individuals. Using previous findings in healthy cells as a reference, we analyzed scRNA-seq data obtained from patients with myelodysplastic syndrome (MDS) and detected specific alterations of the expression dynamics of genes involved in erythroid differentiation in all patients with MDS such as TRIB2. In addition, the comparison between transcriptional programs and GRNs regulating normal HSPCs and MDS HSPCs allowed identification of regulons that were specifically active in MDS cases such as SMAD1, HOXA6, POU2F2, and RUNX1 suggesting a role of these transcription factors (TFs) in the pathogenesis of the disease.
In summary, we demonstrate that the combination of single-cell technologies with computational analysis tools enable the study of a variety of cellular mechanisms involved in complex biological systems such as early hematopoiesis and can be used to dissect perturbed differentiation trajectories associated with perturbations such as aging and malignant transformation. Furthermore, the identification of abnormal regulatory mechanisms associated with myeloid malignancies could be exploited for personalized therapeutic approaches in individual patients.
Keywords: aging; computational biology; genetics; genomics; hematopoietic stem; human; myelodysplastic syndrome; progenitor cells; single-cell RNA sequencing; systems biology.
Plain language summary
Our blood contains many different types of cells; red blood cells carry oxygen through the body, platelets help to stop bleeding and a variety of white blood cells fight infections. All of these critical components come from a pool of immature cells in bone marrow, which can develop and specialise into any of these. However, as we get older, these immature cells can accumulate damage, including mutations in specific genes. This increases the risk of diseases such as myelodysplastic syndromes (MDS), a type of cancer in which the cells cannot develop and the patient does not have enough healthy mature blood cells. The changes in gene activity in the immature cells have previously been studied using samples from young and elderly people, as well as individuals with MDS. These studies examined large numbers of cells together, revealing differences between young and elderly people, and individuals with MDS. However, this does not describe how the different types alter their behaviour. To address this, Ainciburu, Ezponda et al. used a technique called single-cell RNA sequencing to study the gene activity in individual immature blood cells. This revealed changes associated with maturation that may account for the different combinations of cell populations in younger and older people. The results confirmed findings from previous studies and suggested new genes involved in ageing or MDS. Ainciburu, Ezponda et al. used these results to create an analytical system that highlights gene activity differences in individual MDS patients that are independent of age-related changes. These results provide new insights that could help further research into the development of MDS and the ageing process. In addition, scientists could study other diseases using this approach of analysing individual patients’ gene activity. In future, this could help to personalise clinical decisions on diagnosis and treatment.
© 2023, Ainciburu, Ezponda et al.
Conflict of interest statement
MA, TE, NB, AA, AV, PS, DA, JL, MS, TJ, FL, SM, FS, AM, JM, GS, AD, ML, DG, MD, DV, MH, FP No competing interests declared, JR Employed by 10x Genomics since February 2021; this employment had no bearing on this work
Figures

References
-
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S. Scenic: single-cell regulatory network inference and clustering. Nature Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
