

A novel fold for acyltransferase-3 (AT3) proteins provides a framework for transmembrane acyl-group transfer
- PMID: 36630168
- PMCID: PMC9833829
- DOI: 10.7554/eLife.81547
A novel fold for acyltransferase-3 (AT3) proteins provides a framework for transmembrane acyl-group transfer
Abstract
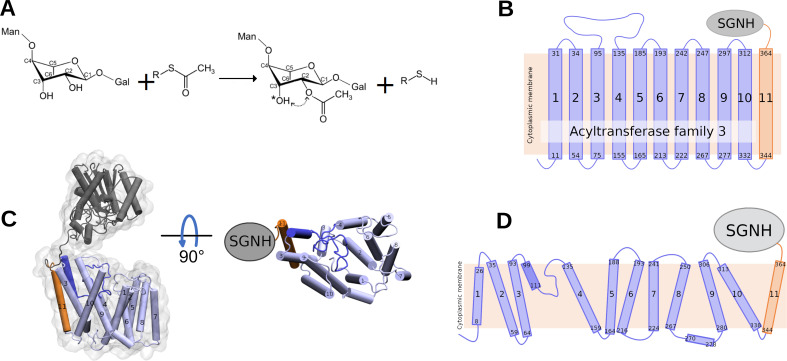
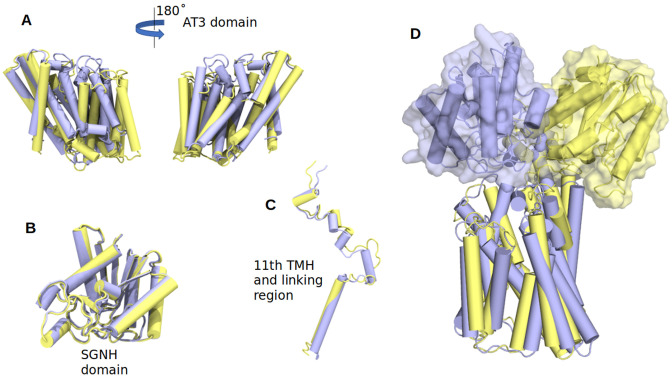
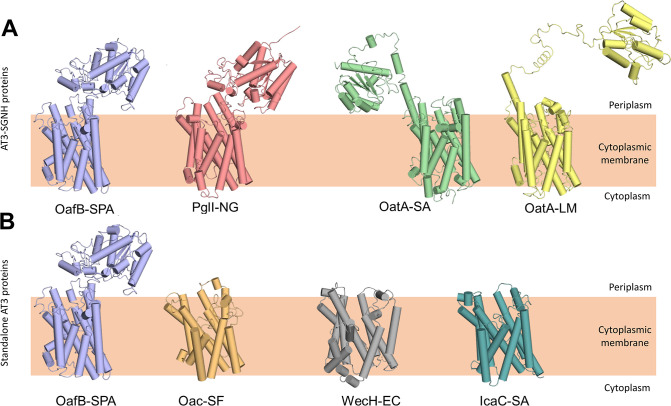
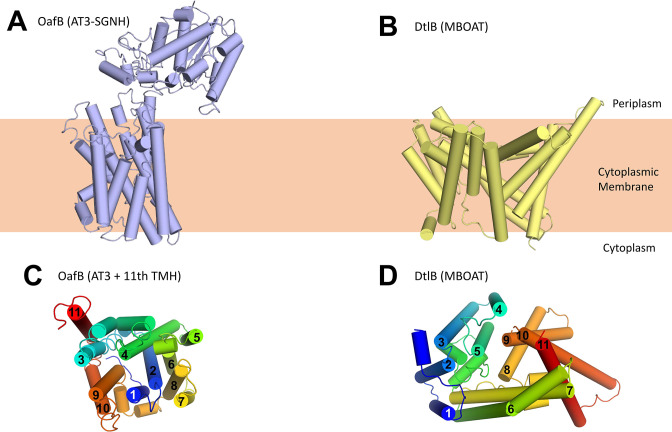
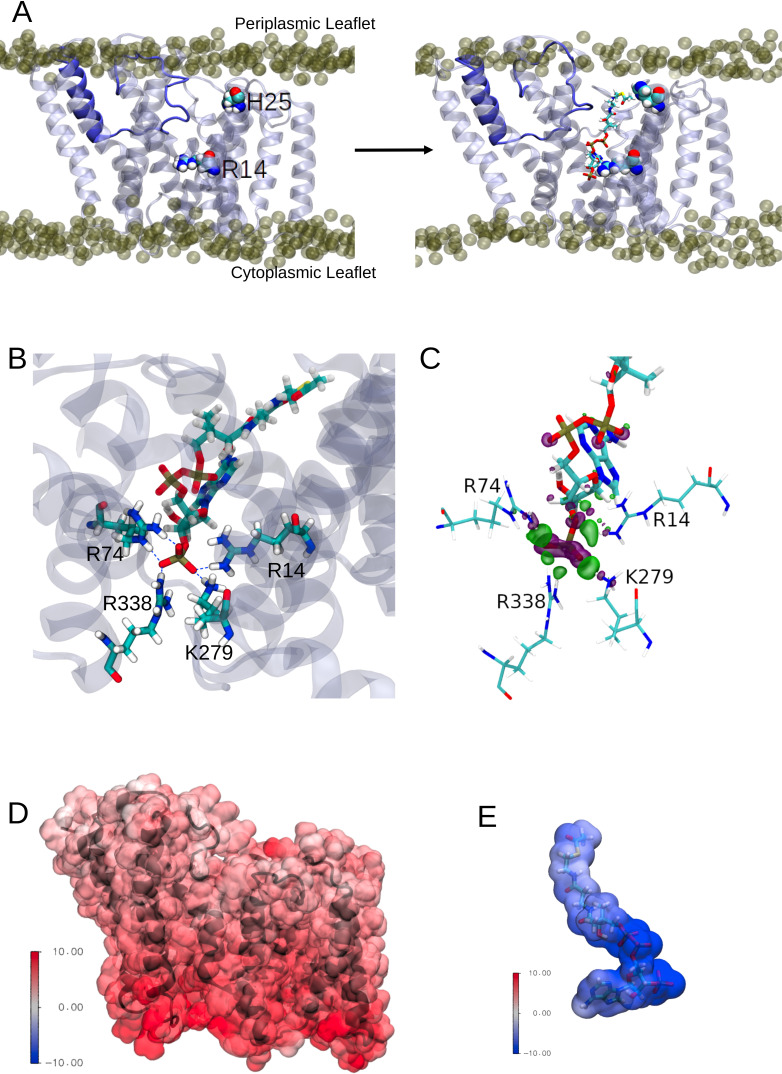
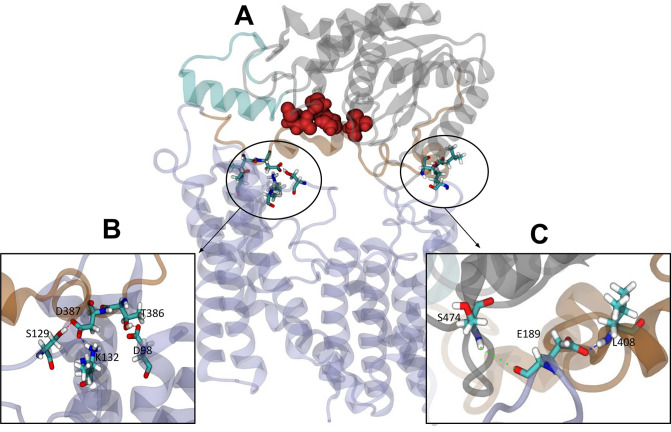
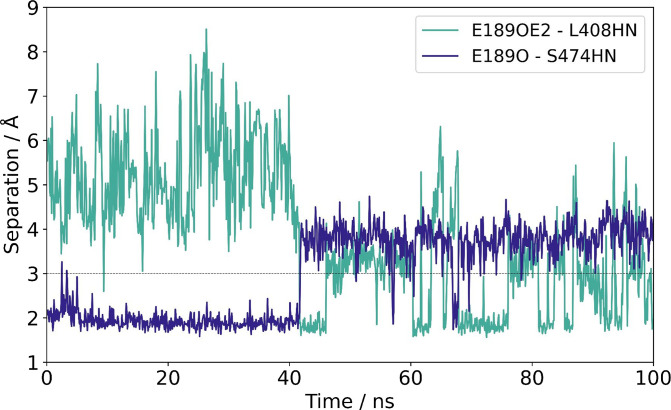
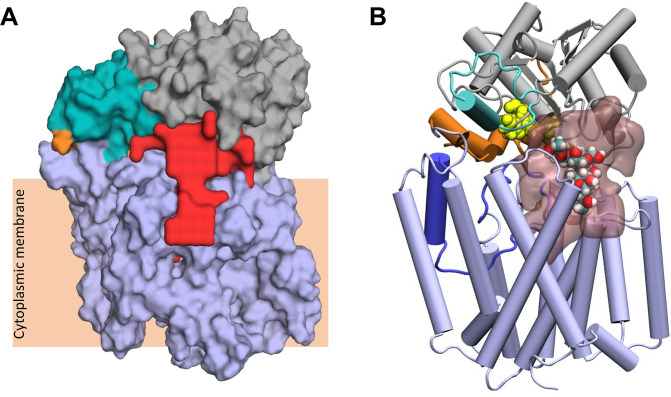
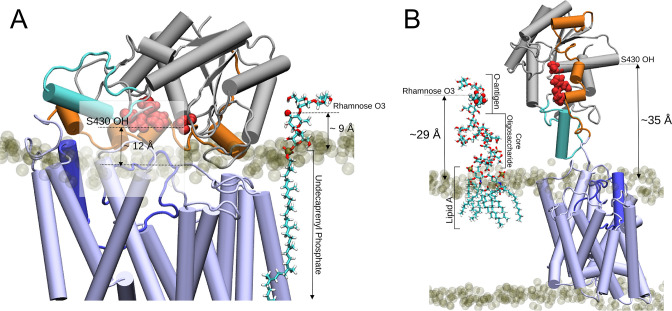
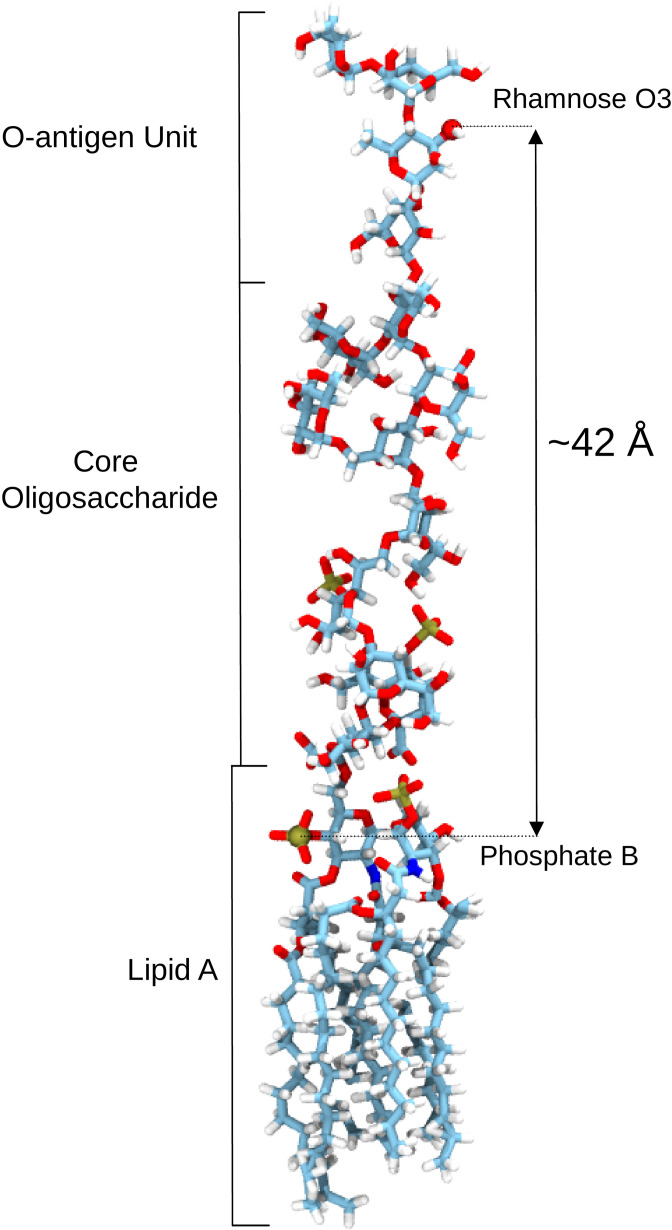
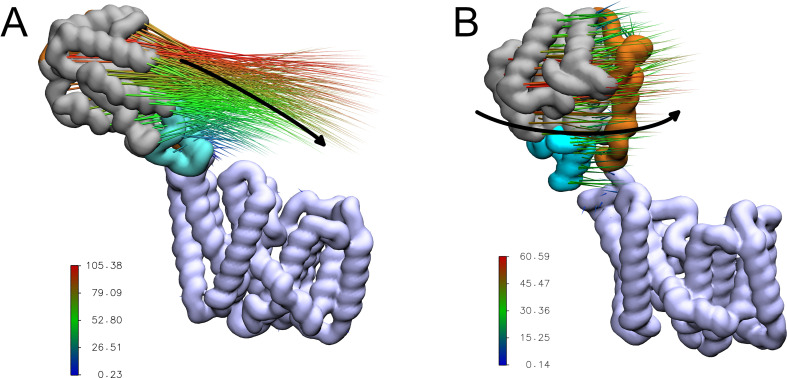
Acylation of diverse carbohydrates occurs across all domains of life and can be catalysed by proteins with a membrane bound acyltransferase-3 (AT3) domain (PF01757). In bacteria, these proteins are essential in processes including symbiosis, resistance to viruses and antimicrobials, and biosynthesis of antibiotics, yet their structure and mechanism are largely unknown. In this study, evolutionary co-variance analysis was used to build a computational model of the structure of a bacterial O-antigen modifying acetyltransferase, OafB. The resulting structure exhibited a novel fold for the AT3 domain, which molecular dynamics simulations demonstrated is stable in the membrane. The AT3 domain contains 10 transmembrane helices arranged to form a large cytoplasmic cavity lined by residues known to be essential for function. Further molecular dynamics simulations support a model where the acyl-coA donor spans the membrane through accessing a pore created by movement of an important loop capping the inner cavity, enabling OafB to present the acetyl group close to the likely catalytic resides on the extracytoplasmic surface. Limited but important interactions with the fused SGNH domain in OafB are identified, and modelling suggests this domain is mobile and can both accept acyl-groups from the AT3 and then reach beyond the membrane to reach acceptor substrates. Together this new general model of AT3 function provides a framework for the development of inhibitors that could abrogate critical functions of bacterial pathogens.
Keywords: Salmonella enterica; acetylation; bacteria; biochemistry; chemical biology; computational biology; lipopolysaccharide; membrane protein; surface polysaccharides; systems biology.
Plain language summary
The fatty membrane that surrounds cells is an essential feature of all living things. It is a selective barrier, only allowing certain substances to enter and exit the cell, and it contains the proteins and carbohydrates that the cell uses to interact with its environment. In bacteria, the carbohydrates on the outer side of the membrane can become ‘tagged’ or modified with small chemical entities which often prove useful for the cell. Acyl groups, for example, allow disease-causing bacteria to evade the immune system and contribute to infections persisting in the body. As a rule, activated acyl groups are only found inside the cell, so they need to move across the membrane before they can be attached onto the carbohydrates at the surface. This transfer is performed by a group of proteins that sit within the membrane called the acyltransferase-3 (AT3) family. The structure of these proteins and the mechanism by which they facilitate membrane crossing have remained unclear. Newman, Tindall et al. combined computational and structural modelling techniques with existing experimental data to establish how this family of proteins moves acyl groups across the membrane. They focused on OafB, an AT3 protein from the foodborne bacterial pathogen Salmonella typhimurium. The experimental data used by the team included information about which parts of OafB are necessary for this protein to acylate carbohydrates molecules. In their experiments, Newman, Tindall et al. studied how different parts of OafB move, how they interact with the molecules that carry an acyl group to the membrane, and how the acyl group is then transferred to the carbohydrate acceptor. Their results suggest that AT3 family proteins have a central pore or hole, plugged by a loop. This loop moves and therefore ‘unplug’ the pore, resulting in the emergence of a channel across the membrane. This channel can accommodate the acyl-donating molecule, presenting the acyl group to the outer surface of the membrane where it can be transferred to the acceptor carbohydrate. The AT3 family of proteins participates in many cellular processes involving the membrane, and a range of bacterial pathogens rely on these proteins to successfully infect human hosts. The results of Newman Tindall et al. could therefore be used across the biological sciences to provide more detailed understanding of the membrane, and to inform the design of drugs to fight bacterial diseases.
© 2023, Newman, Tindall et al.
Conflict of interest statement
KN, ST, SM, SK, GT, MV No competing interests declared
Figures
Similar articles
-
Acyltransferases that Modify Cell Surface Polymers Across the Membrane.Biochemistry. 2025 Apr 15;64(8):1728-1749. doi: 10.1021/acs.biochem.4c00731. Epub 2025 Apr 2. Biochemistry. 2025. PMID: 40171682 Review.
-
Acetylation of Surface Carbohydrates in Bacterial Pathogens Requires Coordinated Action of a Two-Domain Membrane-Bound Acyltransferase.mBio. 2020 Aug 25;11(4):e01364-20. doi: 10.1128/mBio.01364-20. mBio. 2020. PMID: 32843546 Free PMC article.
-
Diverse functions for acyltransferase-3 proteins in the modification of bacterial cell surfaces.Microbiology (Reading). 2022 Mar;168(3):001146. doi: 10.1099/mic.0.001146. Microbiology (Reading). 2022. PMID: 35253642 Free PMC article.
-
The mechanism of peptidoglycan O-acetylation in Gram-negative bacteria typifies bacterial MBOAT-SGNH acyltransferases.bioRxiv [Preprint]. 2024 Sep 19:2024.09.17.613324. doi: 10.1101/2024.09.17.613324. bioRxiv. 2024. Update in: J Biol Chem. 2025 Jun;301(6):108531. doi: 10.1016/j.jbc.2025.108531. PMID: 39345430 Free PMC article. Updated. Preprint.
-
Engineering Aspects of Olfaction.In: Persaud KC, Marco S, Gutiérrez-Gálvez A, editors. Neuromorphic Olfaction. Boca Raton (FL): CRC Press/Taylor & Francis; 2013. Chapter 1. In: Persaud KC, Marco S, Gutiérrez-Gálvez A, editors. Neuromorphic Olfaction. Boca Raton (FL): CRC Press/Taylor & Francis; 2013. Chapter 1. PMID: 26042329 Free Books & Documents. Review.
Cited by
-
Acyltransferases that Modify Cell Surface Polymers Across the Membrane.Biochemistry. 2025 Apr 15;64(8):1728-1749. doi: 10.1021/acs.biochem.4c00731. Epub 2025 Apr 2. Biochemistry. 2025. PMID: 40171682 Review.
-
Structure of the human heparan-α-glucosaminide N-acetyltransferase (HGSNAT).Elife. 2024 Aug 28;13:RP93510. doi: 10.7554/eLife.93510. Elife. 2024. PMID: 39196614 Free PMC article.
-
Phenotypic and Genomic Analysis of Enterobacter ludwigii Strains: Insights into Mechanisms Enhancing Plant Growth Both Under Normal Conditions and in Response to Supplementation with Mineral Fertilizers and Exposure to Stress Factors.Plants (Basel). 2024 Dec 19;13(24):3551. doi: 10.3390/plants13243551. Plants (Basel). 2024. PMID: 39771249 Free PMC article.
-
Structure of the human heparan-α-glucosaminide N-acetyltransferase (HGSNAT).bioRxiv [Preprint]. 2024 Jun 12:2023.10.23.563672. doi: 10.1101/2023.10.23.563672. bioRxiv. 2024. Update in: Elife. 2024 Aug 28;13:RP93510. doi: 10.7554/eLife.93510. PMID: 37961489 Free PMC article. Updated. Preprint.
References
-
- Arisawa A, Kawamura N, Takeda K, Tsunekawa H, Okamura K, Okamoto R. Cloning of the macrolide antibiotic biosynthesis gene acya, which encodes 3-O-acyltransferase, from Streptomyces thermotolerans and its use for direct fermentative production of a hybrid macrolide antibiotic. Applied and Environmental Microbiology. 1994;60:2657–2660. doi: 10.1128/aem.60.7.2657-2660.1994. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
