

Innate immune evasion strategies of SARS-CoV-2
- PMID: 36631691
- PMCID: PMC9838430
- DOI: 10.1038/s41579-022-00839-1
Innate immune evasion strategies of SARS-CoV-2
Abstract
SARS-CoV-2, the virus responsible for the COVID-19 pandemic, has been associated with substantial global morbidity and mortality. Despite a tropism that is largely confined to the airways, COVID-19 is associated with multiorgan dysfunction and long-term cognitive pathologies. A major driver of this biology stems from the combined effects of virus-mediated interference with the host antiviral defences in infected cells and the sensing of pathogen-associated material by bystander cells. Such a dynamic results in delayed induction of type I and III interferons (IFN-I and IFN-III) at the site of infection, but systemic IFN-I and IFN-III priming in distal organs and barrier epithelial surfaces, respectively. In this Review, we examine the relationship between SARS-CoV-2 biology and the cellular response to infection, detailing how antagonism and dysregulation of host innate immune defences contribute to disease severity of COVID-19.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
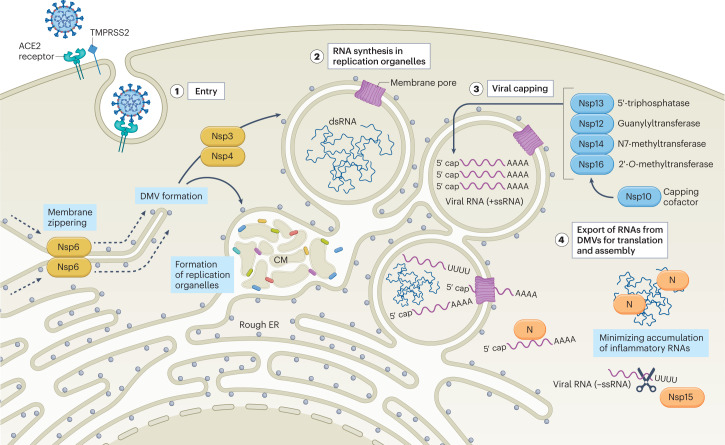
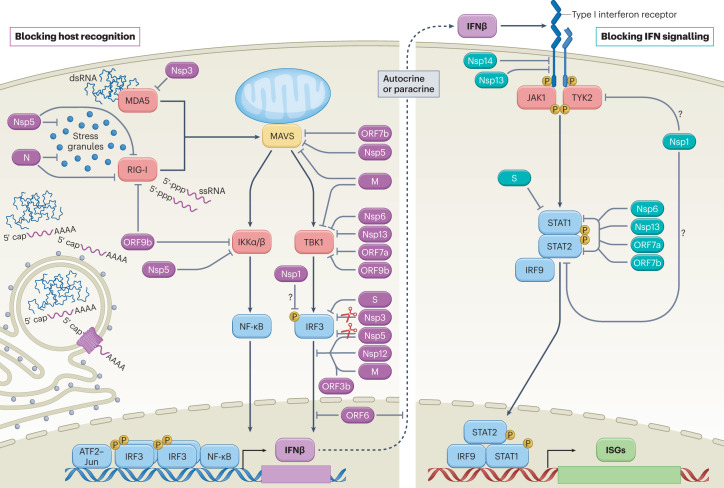
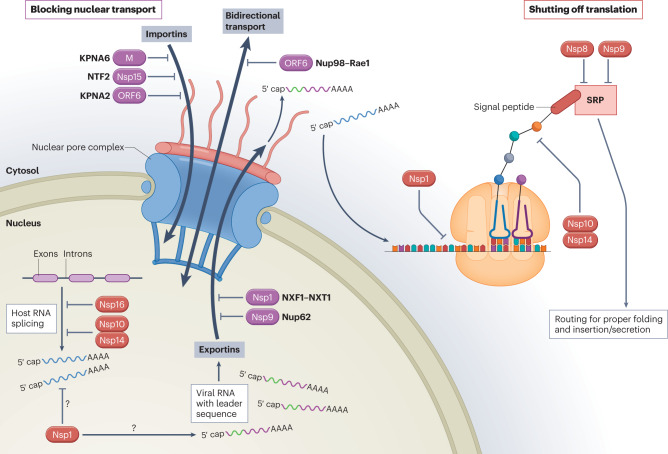
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
