

Association of Genetic Diagnoses for Childhood-Onset Hearing Loss With Cochlear Implant Outcomes
- PMID: 36633841
- PMCID: PMC9857764
- DOI: 10.1001/jamaoto.2022.4463
Association of Genetic Diagnoses for Childhood-Onset Hearing Loss With Cochlear Implant Outcomes
Abstract
Importance: In the US, most childhood-onset bilateral sensorineural hearing loss is genetic, with more than 120 genes and thousands of different alleles known. Primary treatments are hearing aids and cochlear implants. Genetic diagnosis can inform progression of hearing loss, indicate potential syndromic features, and suggest best timing for individualized treatment.
Objective: To identify the genetic causes of childhood-onset hearing loss and characterize severity, progression, and cochlear implant success associated with genotype in a single large clinical cohort.
Design, setting, and participants: This cross-sectional analysis (genomics) and retrospective cohort analysis (audiological measures) were conducted from 2019 to 2022 at the otolaryngology and audiology clinics of Seattle Children's Hospital and the University of Washington and included 449 children from 406 families with bilateral sensorineural hearing loss with an onset younger than 18 years. Data were analyzed between January and June 2022.
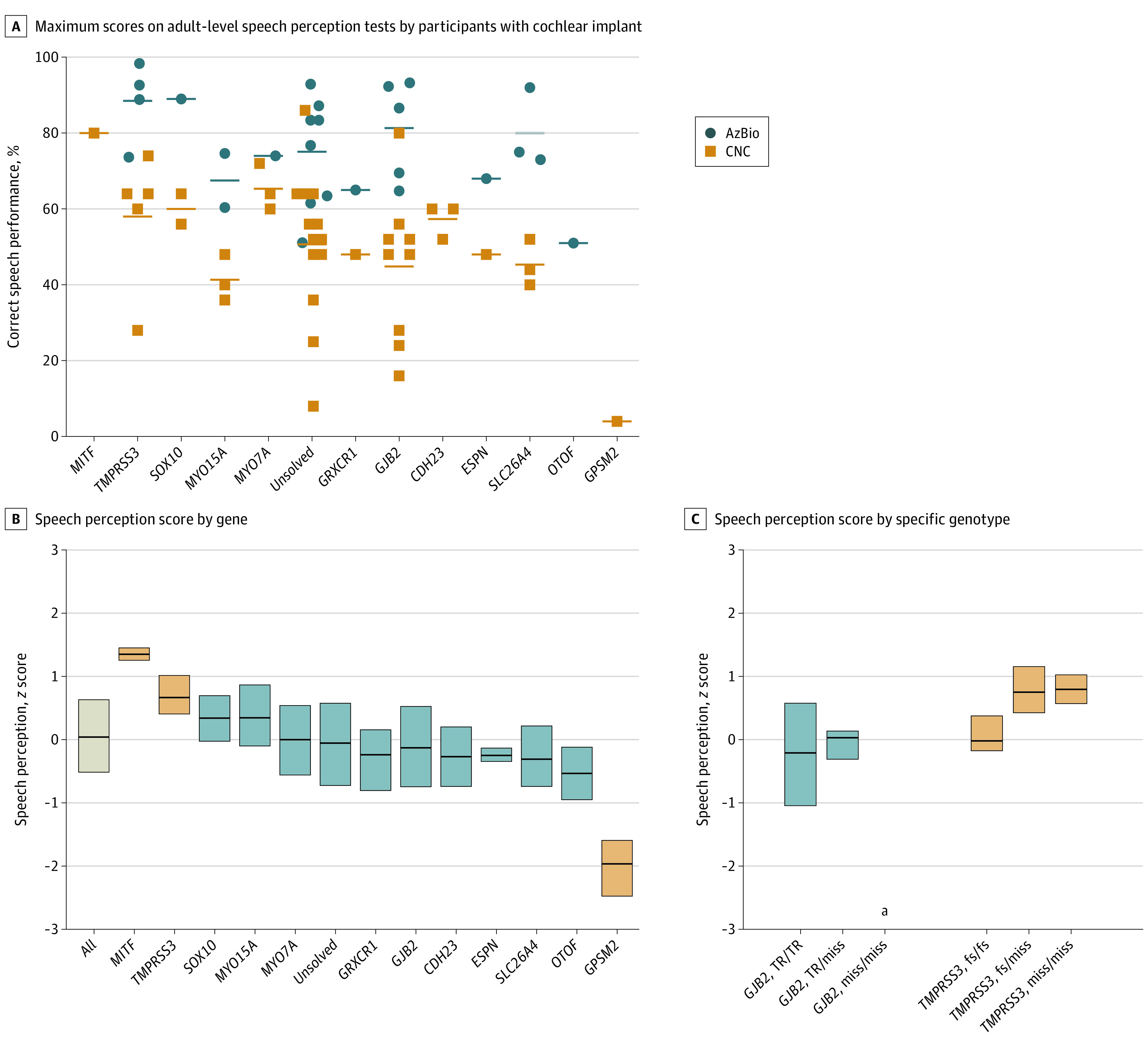
Main outcomes and measures: Genetic diagnoses based on genomic sequencing and structural variant analysis of the DNA of participants; severity and progression of hearing loss as measured by audiologic testing; and cochlear implant success as measured by pediatric and adult speech perception tests. Hearing thresholds and speech perception scores were evaluated with respect to age at implant, months since implant, and genotype using a multivariate analysis of variance and covariance.
Results: Of 406 participants, 208 (51%) were female, 17 (4%) were African/African American, 32 (8%) were East Asian, 219 (54%) were European, 53 (13%) were Latino/Admixed American, and 16 (4%) were South Asian. Genomic analysis yielded genetic diagnoses for 210 of 406 families (52%), including 55 of 82 multiplex families (67%) and 155 of 324 singleton families (48%). Rates of genetic diagnosis were similar for children of all ancestries. Causal variants occurred in 43 different genes, with each child (with 1 exception) having causative variant(s) in only 1 gene. Hearing loss severity, affected frequencies, and progression varied by gene and, for some genes, by genotype within gene. For children with causative mutations in MYO6, OTOA, SLC26A4, TMPRSS3, or severe loss-of-function variants in GJB2, hearing loss was progressive, with losses of more than 10 dB per decade. For all children with cochlear implants, outcomes of adult speech perception tests were greater than preimplanted levels. Yet the degree of success varied substantially by genotype. Adjusting for age at implant and interval since implant, speech perception was highest for children with hearing loss due to MITF or TMPRSS3.
Conclusions and relevance: The results of this cross-sectional study suggest that genetic diagnosis is now sufficiently advanced to enable its integration into precision medical care for childhood-onset hearing loss.
Conflict of interest statement
Figures



References
-
- Van Camp G, Smith R. Hereditary hearing loss homepage. Accessed August 31, 2022. https://hereditaryhearingloss.org.
-
- Shearer EA, Hildebrand MS, Smith RJ. Gene reviews. In: Adam M, Ardinger H, Pagon R, eds. Gene Reviews. National Institutes of Health; 2017.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
