

Reduced methylation correlates with diabetic nephropathy risk in type 1 diabetes
- PMID: 36633903
- PMCID: PMC9927943
- DOI: 10.1172/JCI160959
Reduced methylation correlates with diabetic nephropathy risk in type 1 diabetes
Abstract
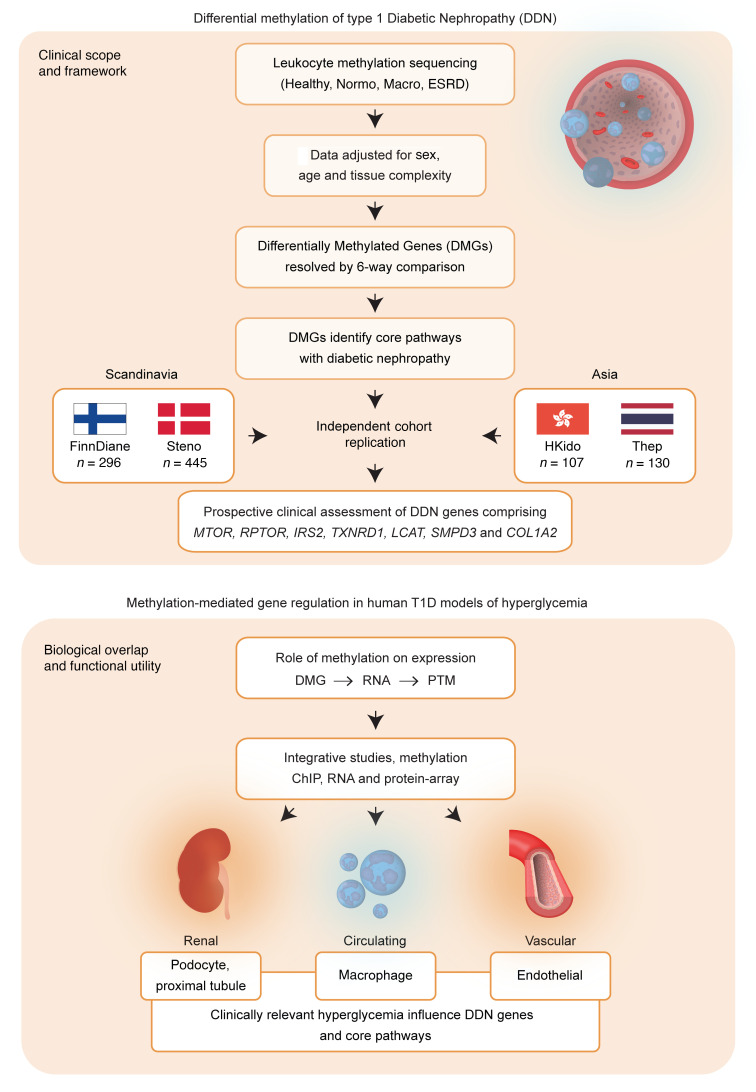
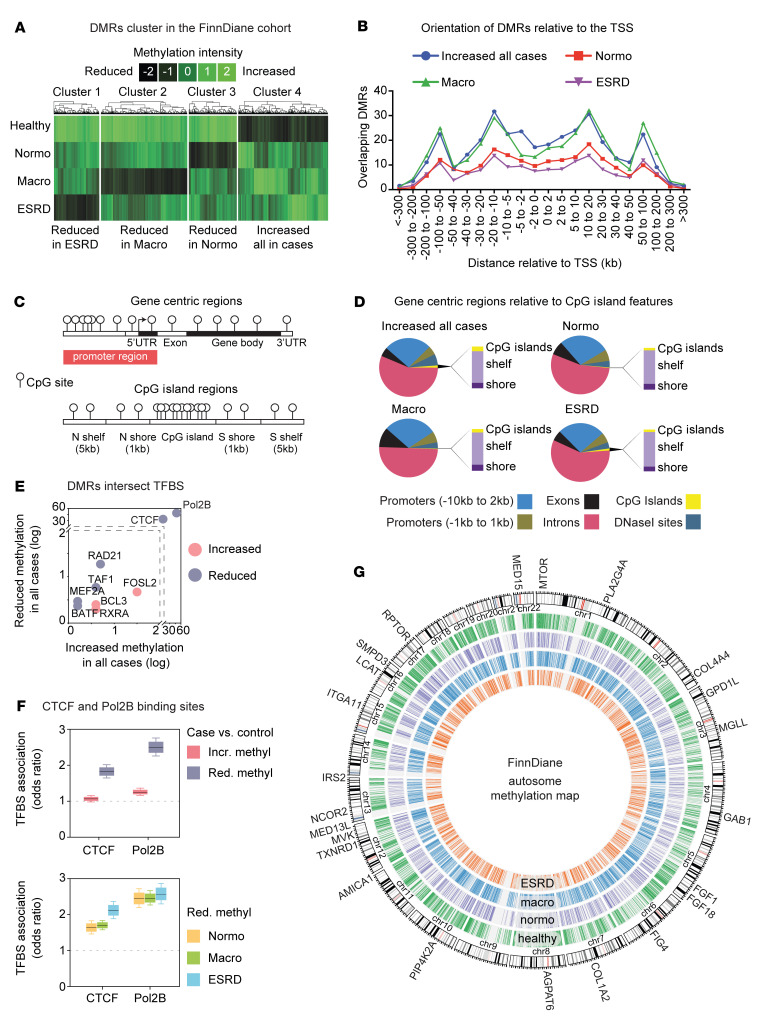
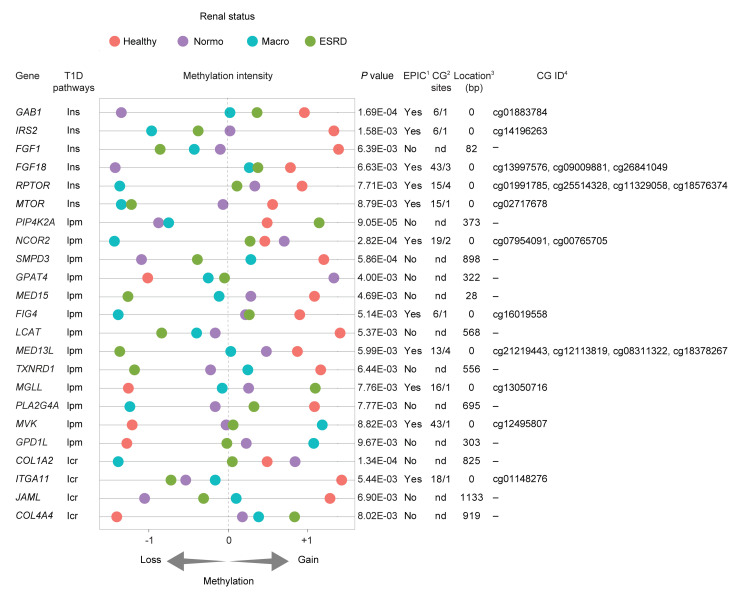
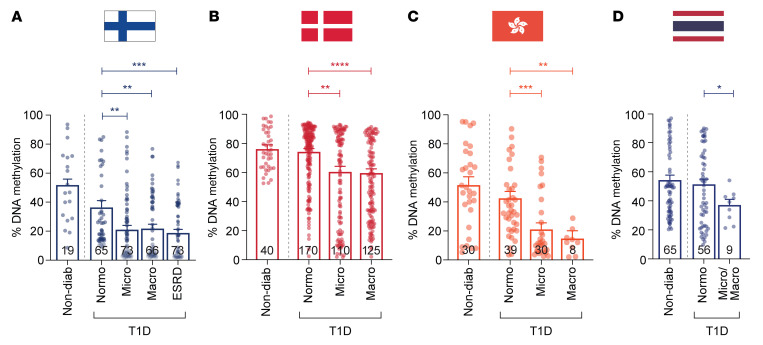
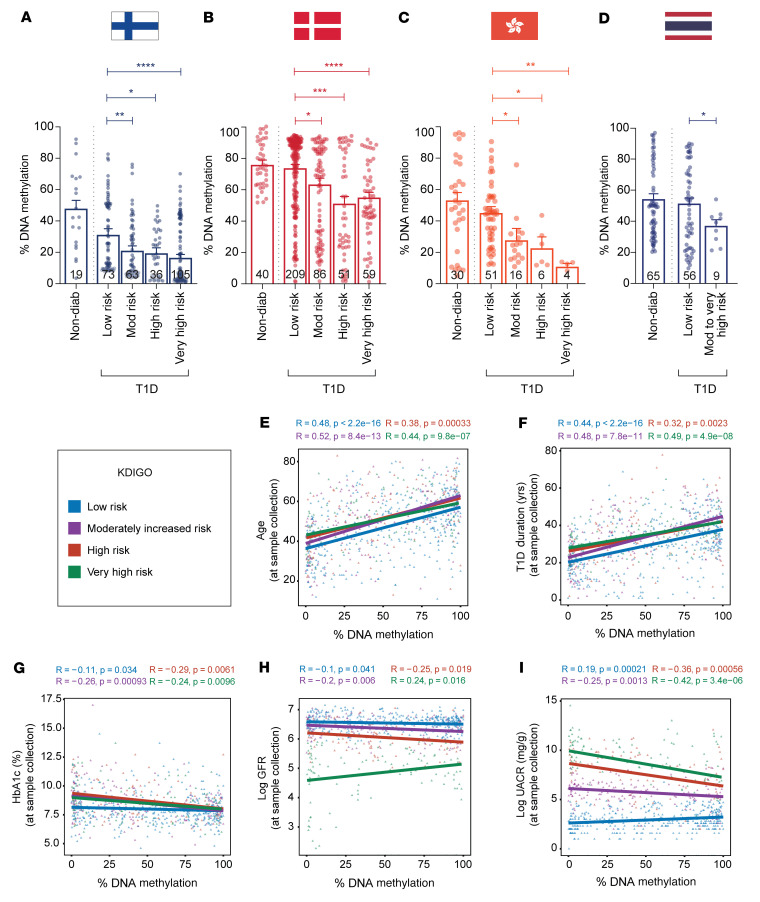
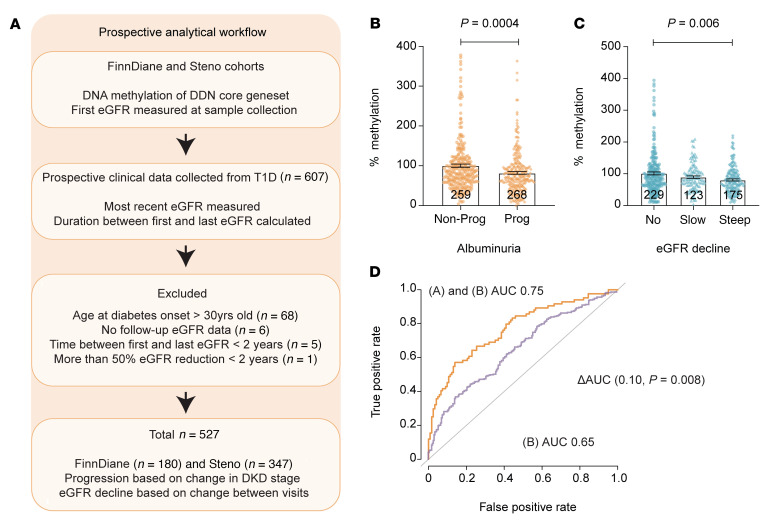
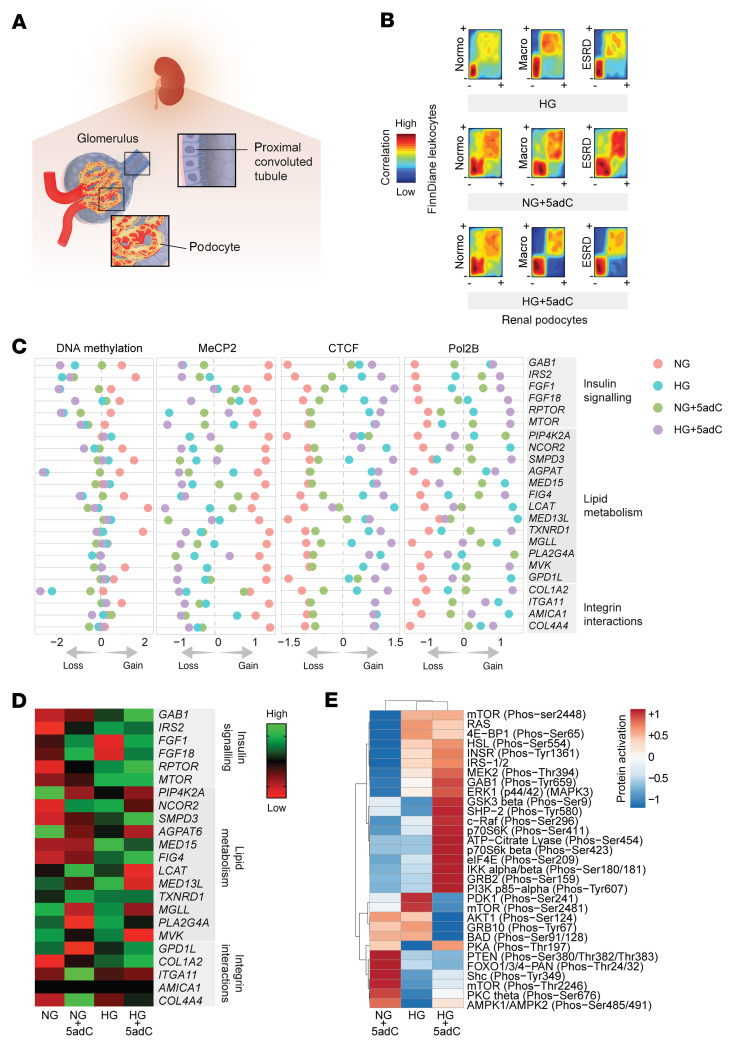
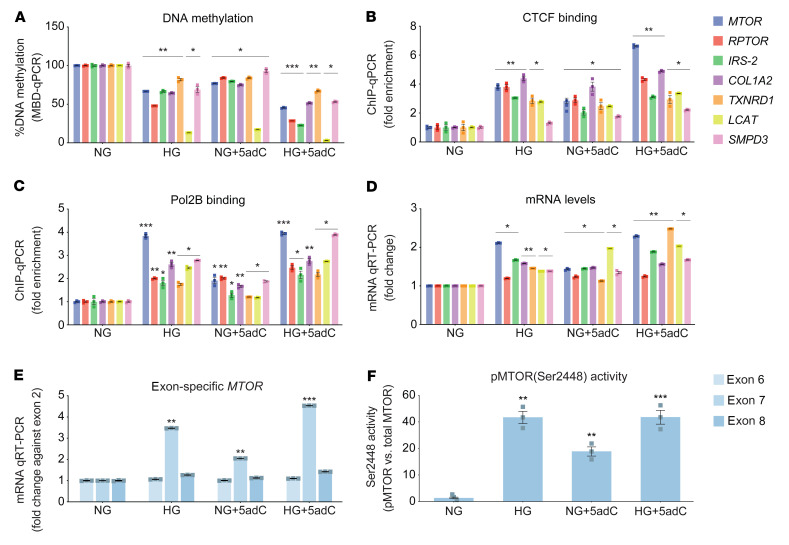
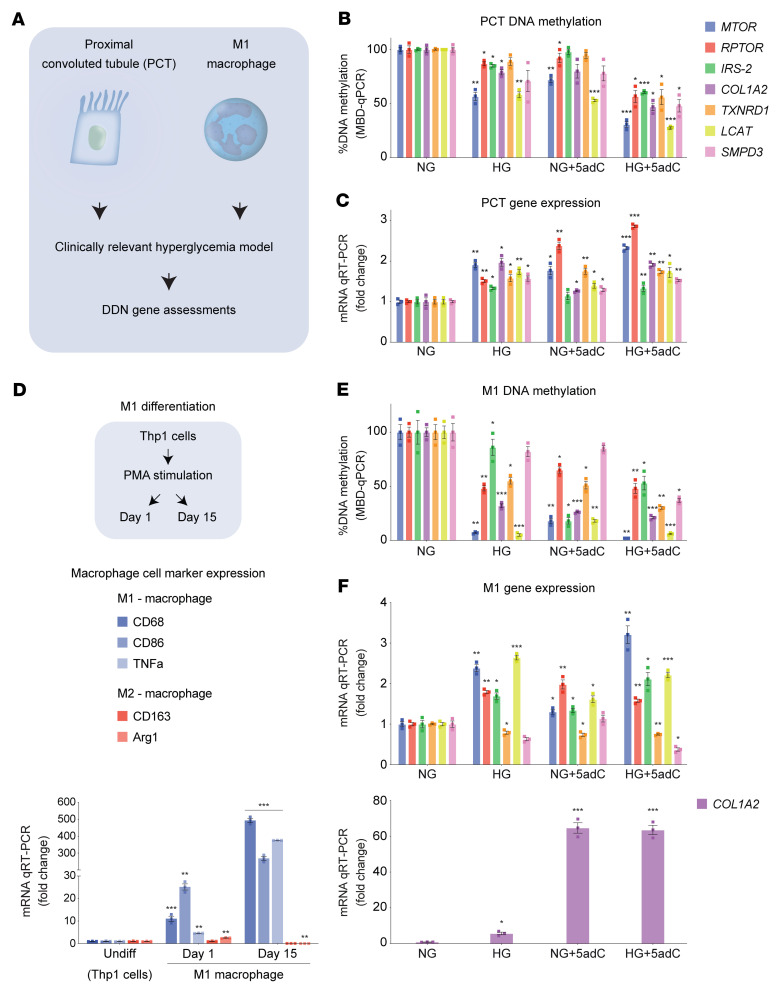
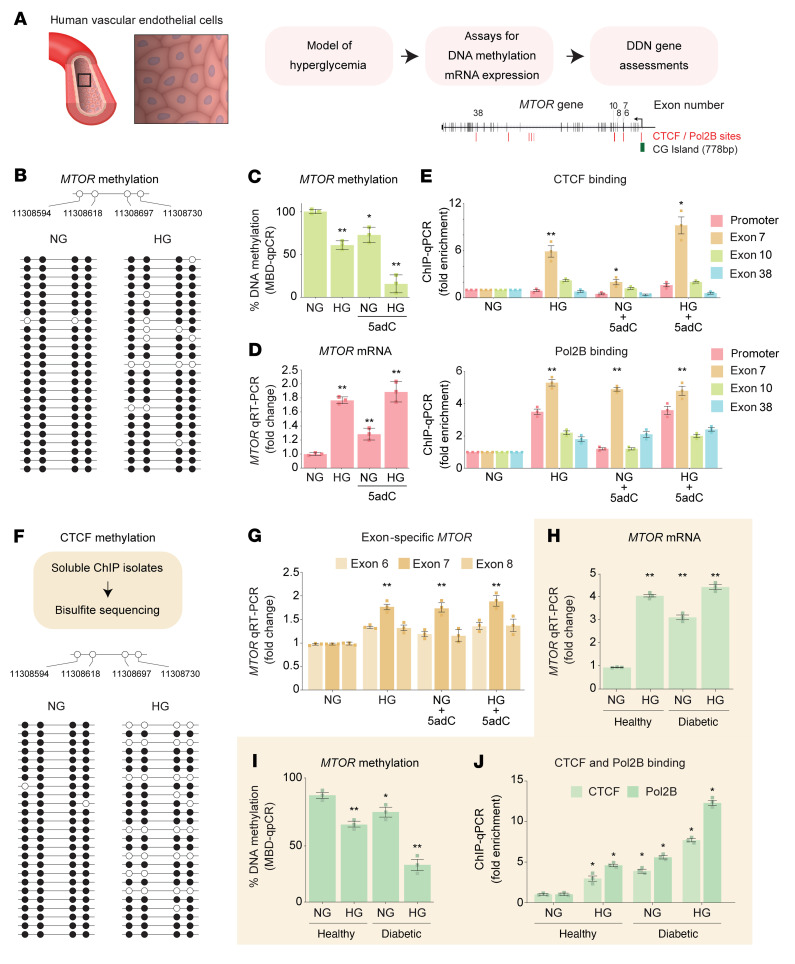
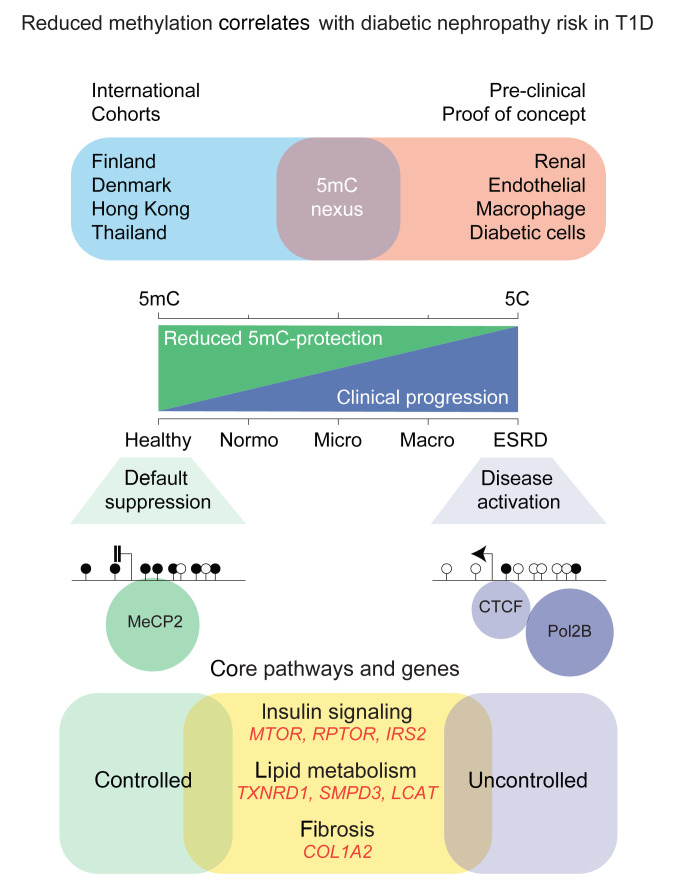
Diabetic nephropathy (DN) is a polygenic disorder with few risk variants showing robust replication in large-scale genome-wide association studies. To understand the role of DNA methylation, it is important to have the prevailing genomic view to distinguish key sequence elements that influence gene expression. This is particularly challenging for DN because genome-wide methylation patterns are poorly defined. While methylation is known to alter gene expression, the importance of this causal relationship is obscured by array-based technologies since coverage outside promoter regions is low. To overcome these challenges, we performed methylation sequencing using leukocytes derived from participants of the Finnish Diabetic Nephropathy (FinnDiane) type 1 diabetes (T1D) study (n = 39) that was subsequently replicated in a larger validation cohort (n = 296). Gene body-related regions made up more than 60% of the methylation differences and emphasized the importance of methylation sequencing. We observed differentially methylated genes associated with DN in 3 independent T1D registries originating from Denmark (n = 445), Hong Kong (n = 107), and Thailand (n = 130). Reduced DNA methylation at CTCF and Pol2B sites was tightly connected with DN pathways that include insulin signaling, lipid metabolism, and fibrosis. To define the pathophysiological significance of these population findings, methylation indices were assessed in human renal cells such as podocytes and proximal convoluted tubule cells. The expression of core genes was associated with reduced methylation, elevated CTCF and Pol2B binding, and the activation of insulin-signaling phosphoproteins in hyperglycemic cells. These experimental observations also closely parallel methylation-mediated regulation in human macrophages and vascular endothelial cells.
Keywords: Diabetes; Epigenetics; Metabolism; Nephrology.
Figures
References
-
- de Boer IH, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch Intern Med. 2011;171(5):412–420. doi: 10.1001/archinternmed.2011.16. - DOI - PMC - PubMed
-
- Diabetes C, et al. Modern-day clinical course of type 1 diabetes mellitus after 30 years’ duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pittsburgh epidemiology of diabetes complications experience (1983-2005) Arch Intern Med. 2009;169(14):1307–1316. doi: 10.1001/archinternmed.2009.193. - DOI - PMC - PubMed
