

Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development
- PMID: 36635388
- PMCID: PMC9961399
- DOI: 10.1038/s41588-022-01276-9
Comprehensive multi-omic profiling of somatic mutations in malformations of cortical development
Abstract
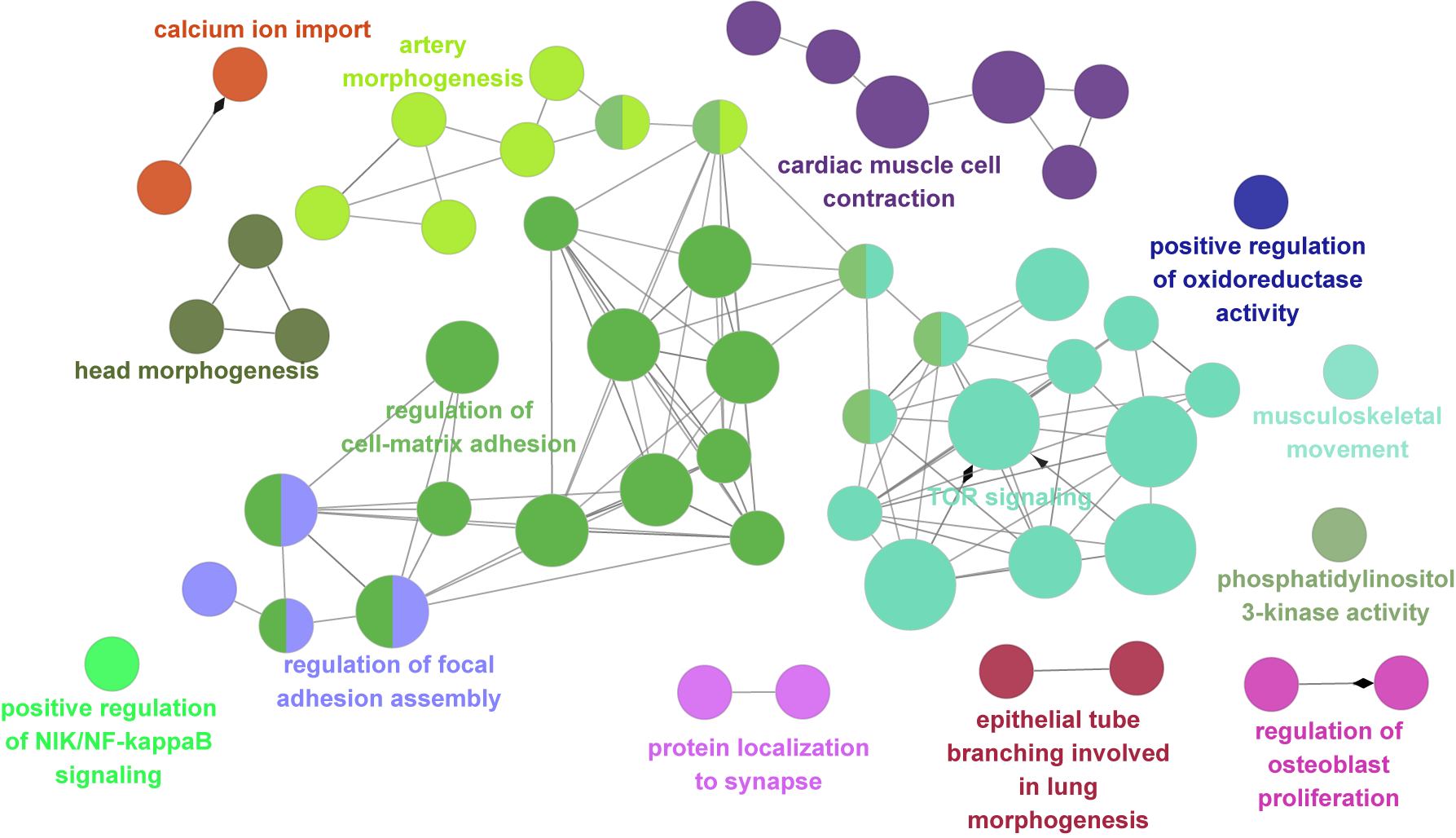
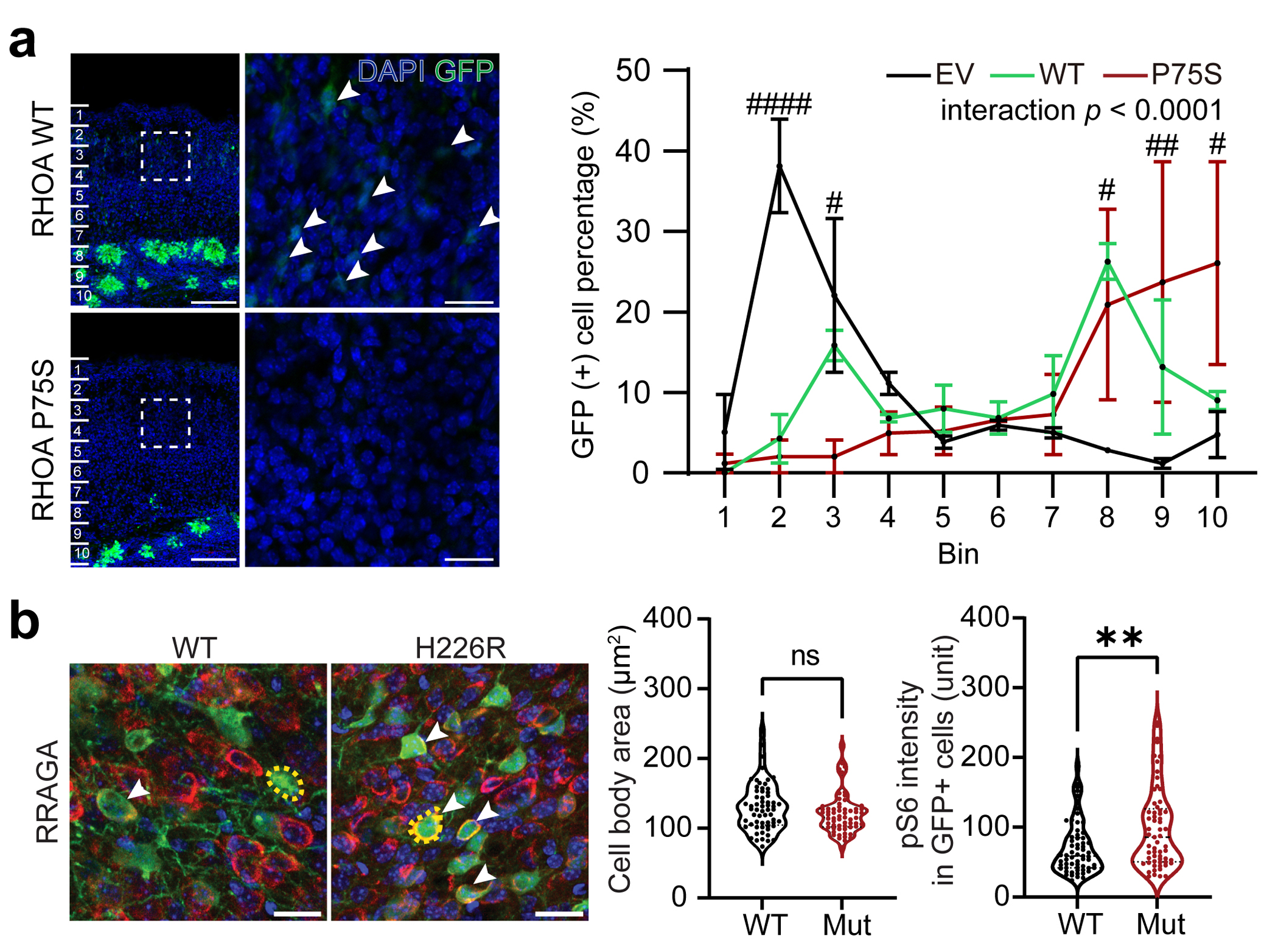
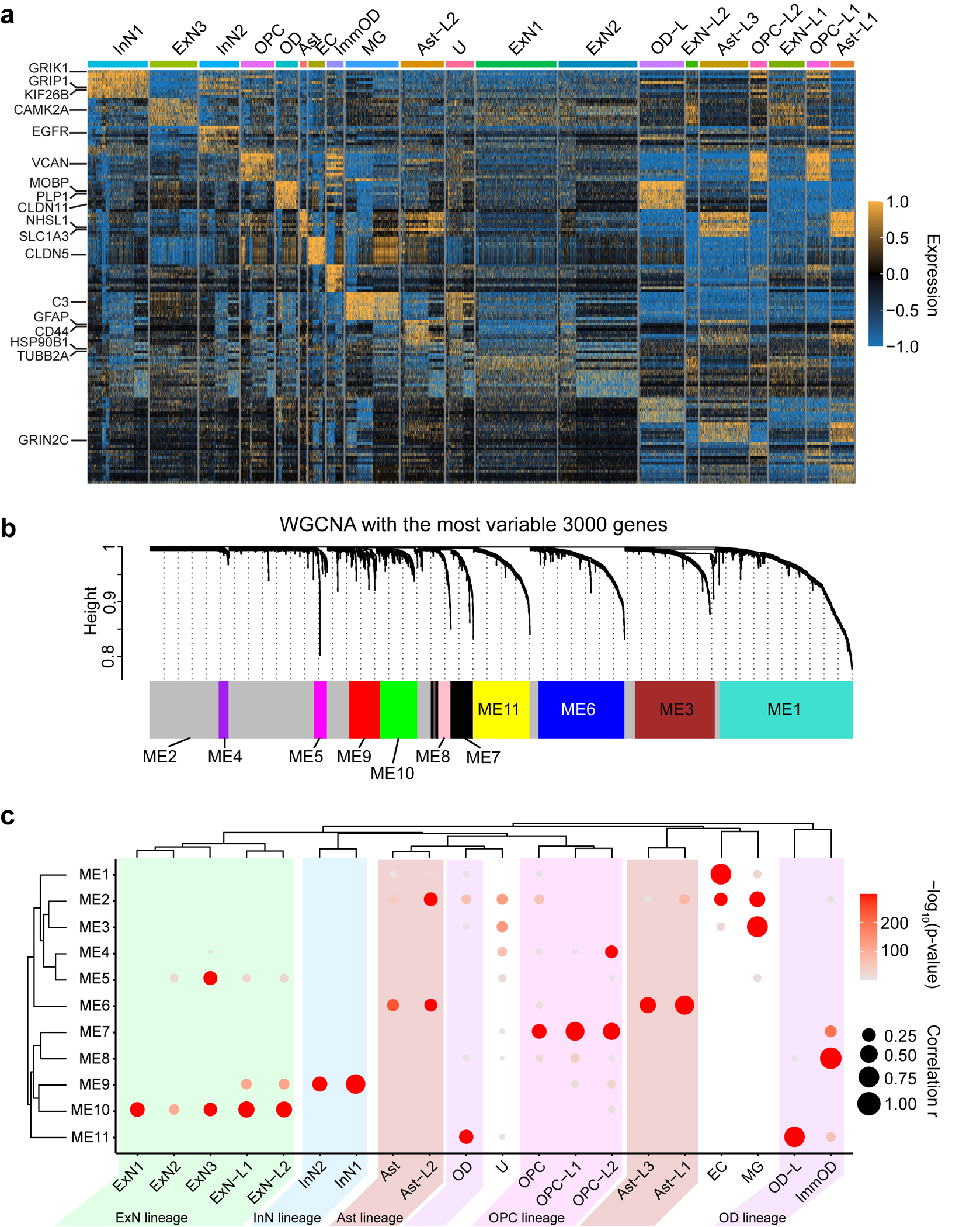
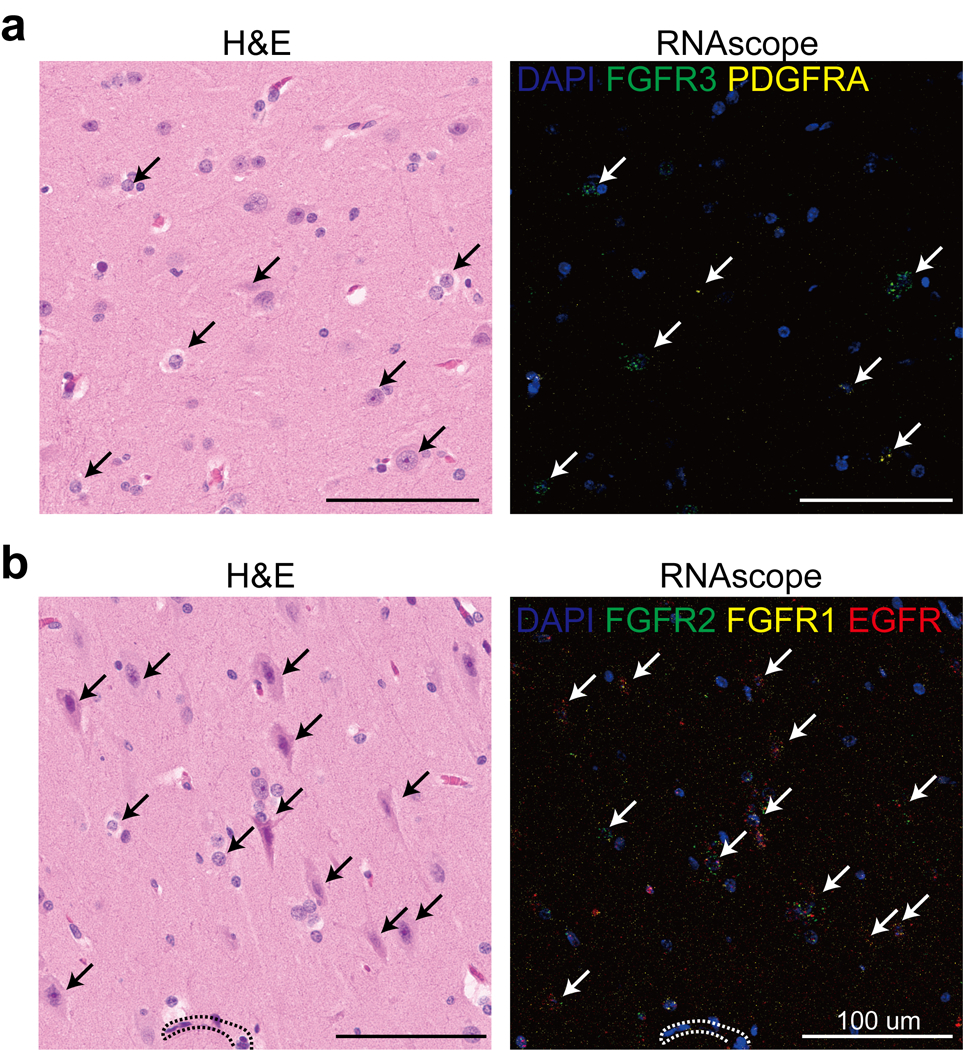
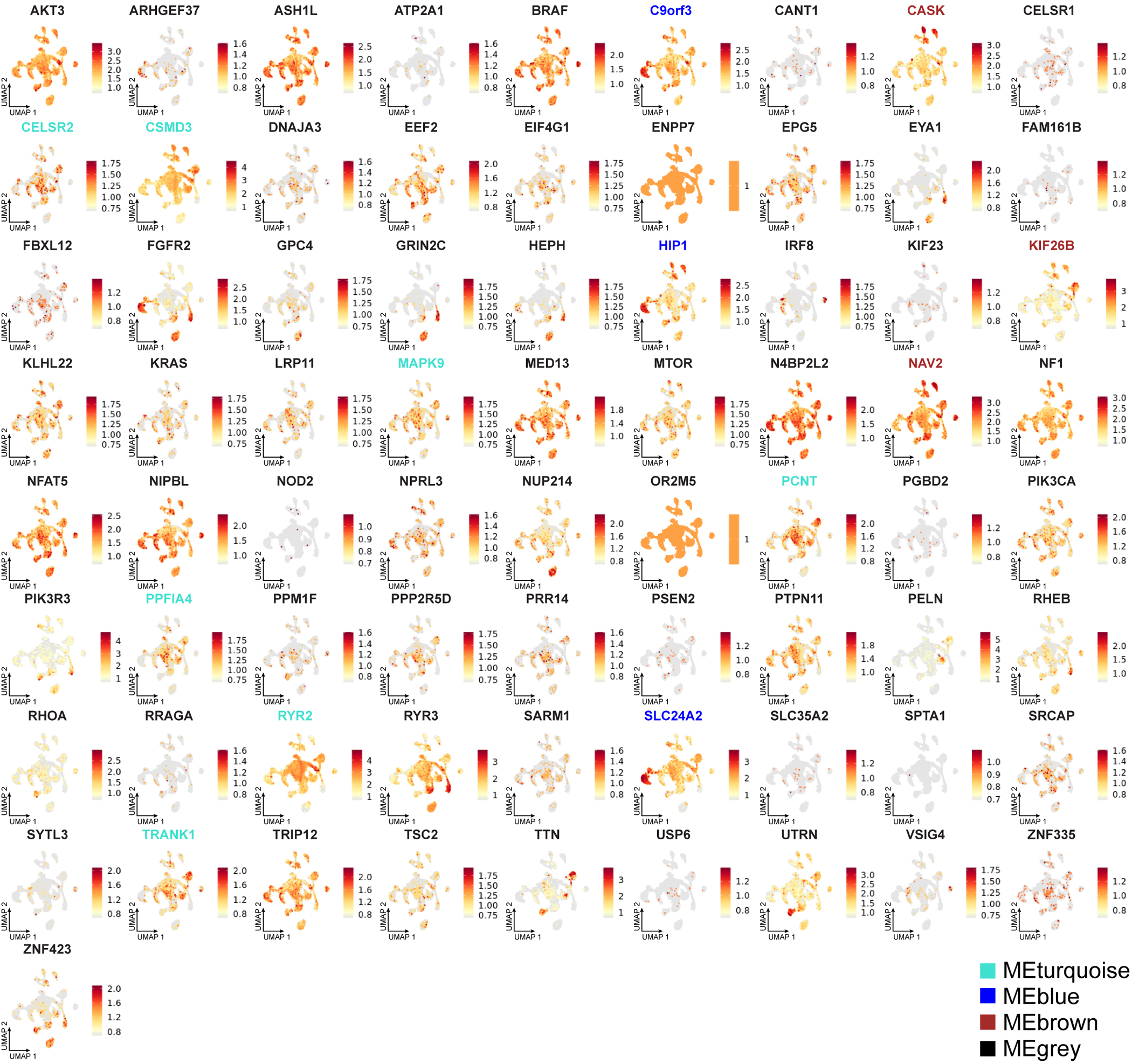
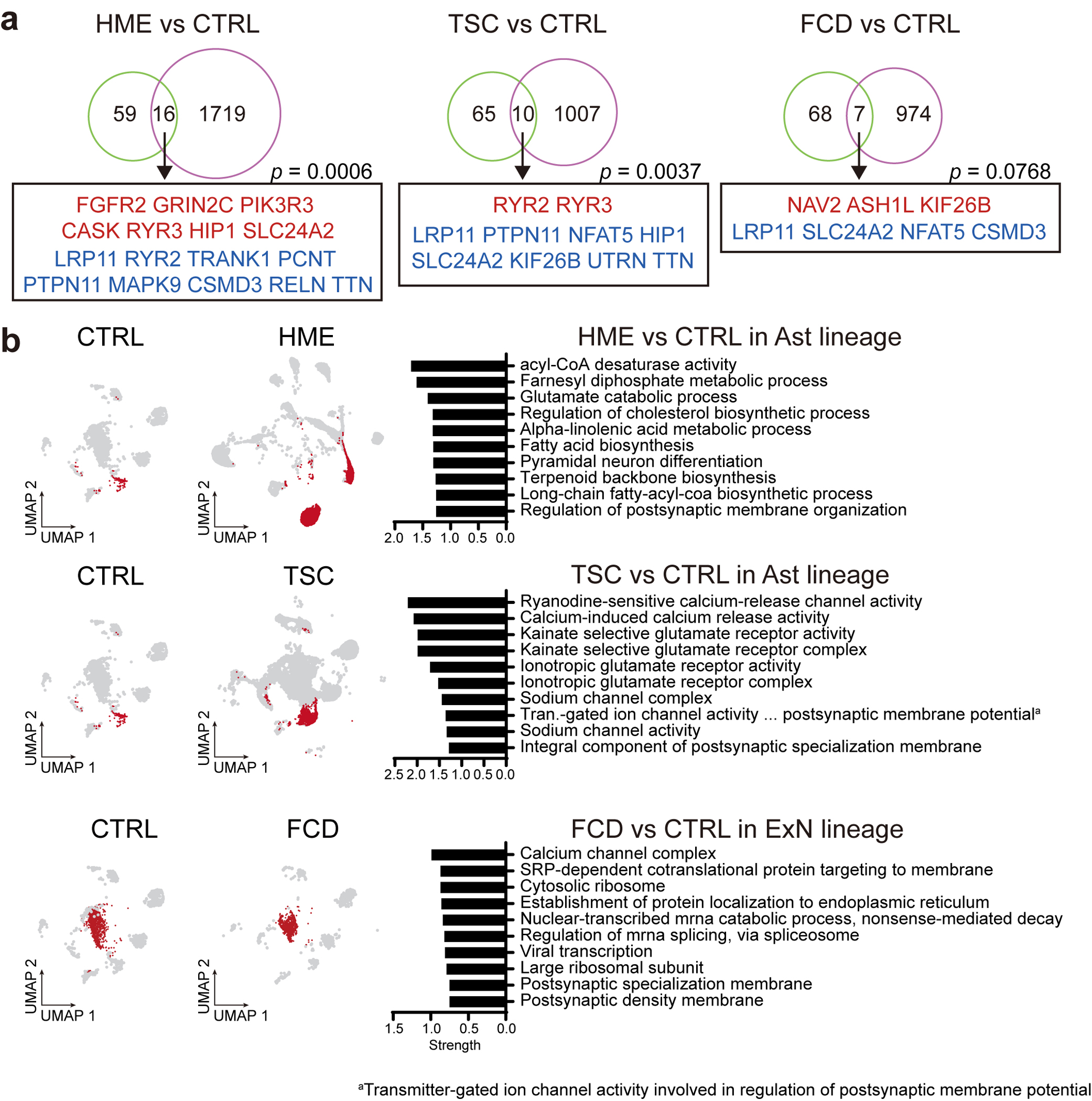
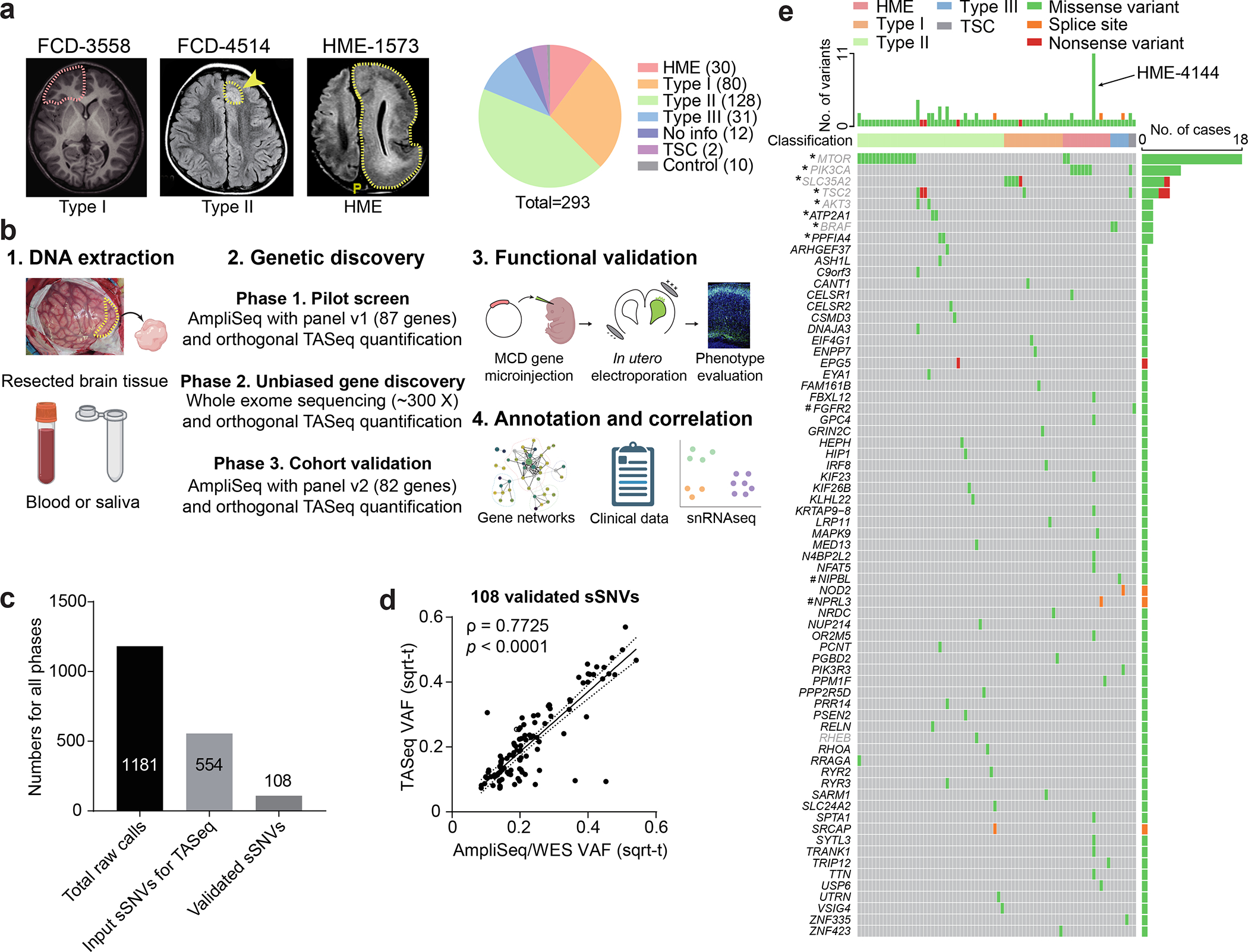
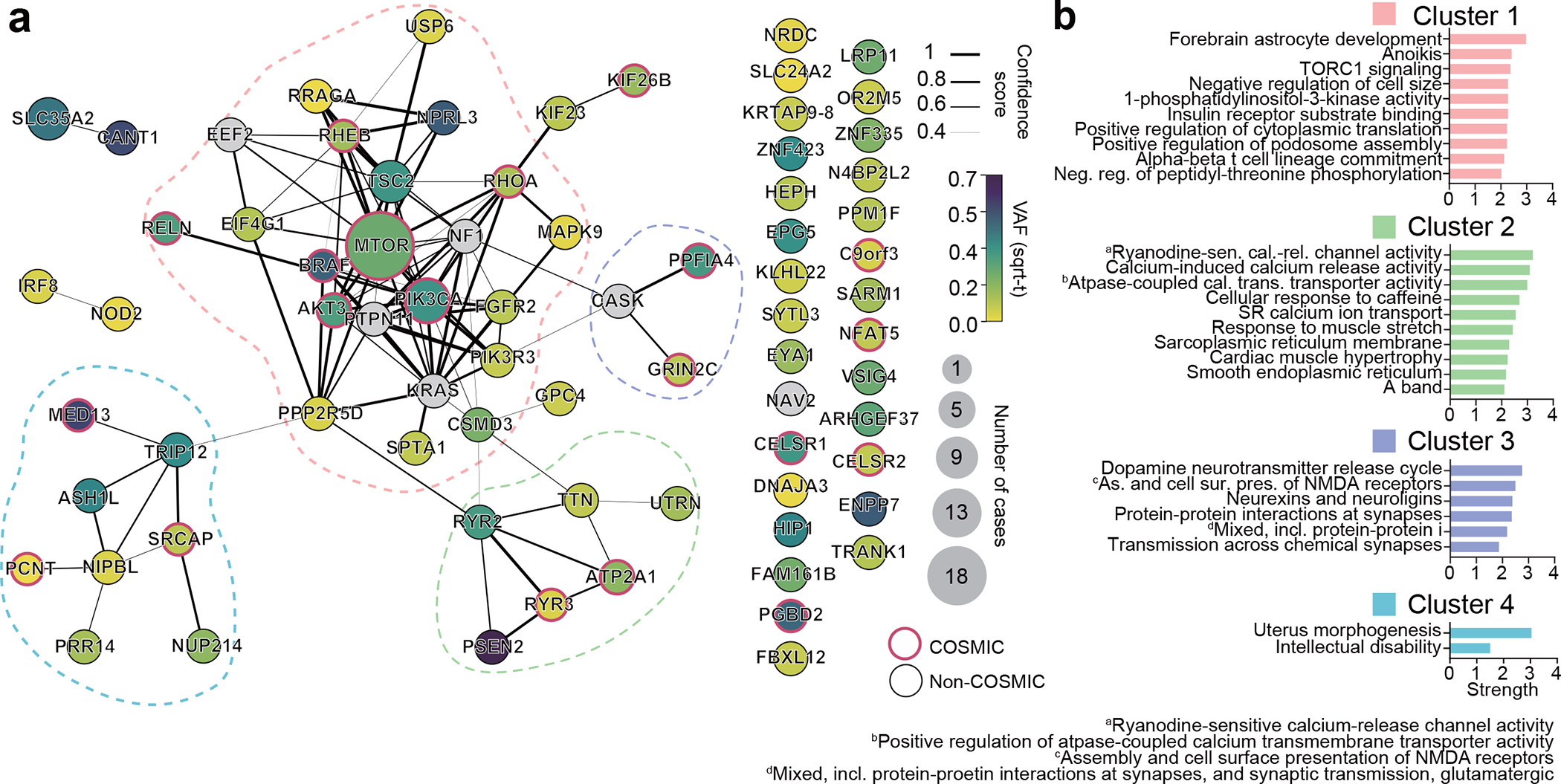
Malformations of cortical development (MCD) are neurological conditions involving focal disruptions of cortical architecture and cellular organization that arise during embryogenesis, largely from somatic mosaic mutations, and cause intractable epilepsy. Identifying the genetic causes of MCD has been a challenge, as mutations remain at low allelic fractions in brain tissue resected to treat condition-related epilepsy. Here we report a genetic landscape from 283 brain resections, identifying 69 mutated genes through intensive profiling of somatic mutations, combining whole-exome and targeted-amplicon sequencing with functional validation including in utero electroporation of mice and single-nucleus RNA sequencing. Genotype-phenotype correlation analysis elucidated specific MCD gene sets associated with distinct pathophysiological and clinical phenotypes. The unique single-cell level spatiotemporal expression patterns of mutated genes in control and patient brains indicate critical roles in excitatory neurogenic pools during brain development and in promoting neuronal hyperexcitability after birth.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests.
Figures
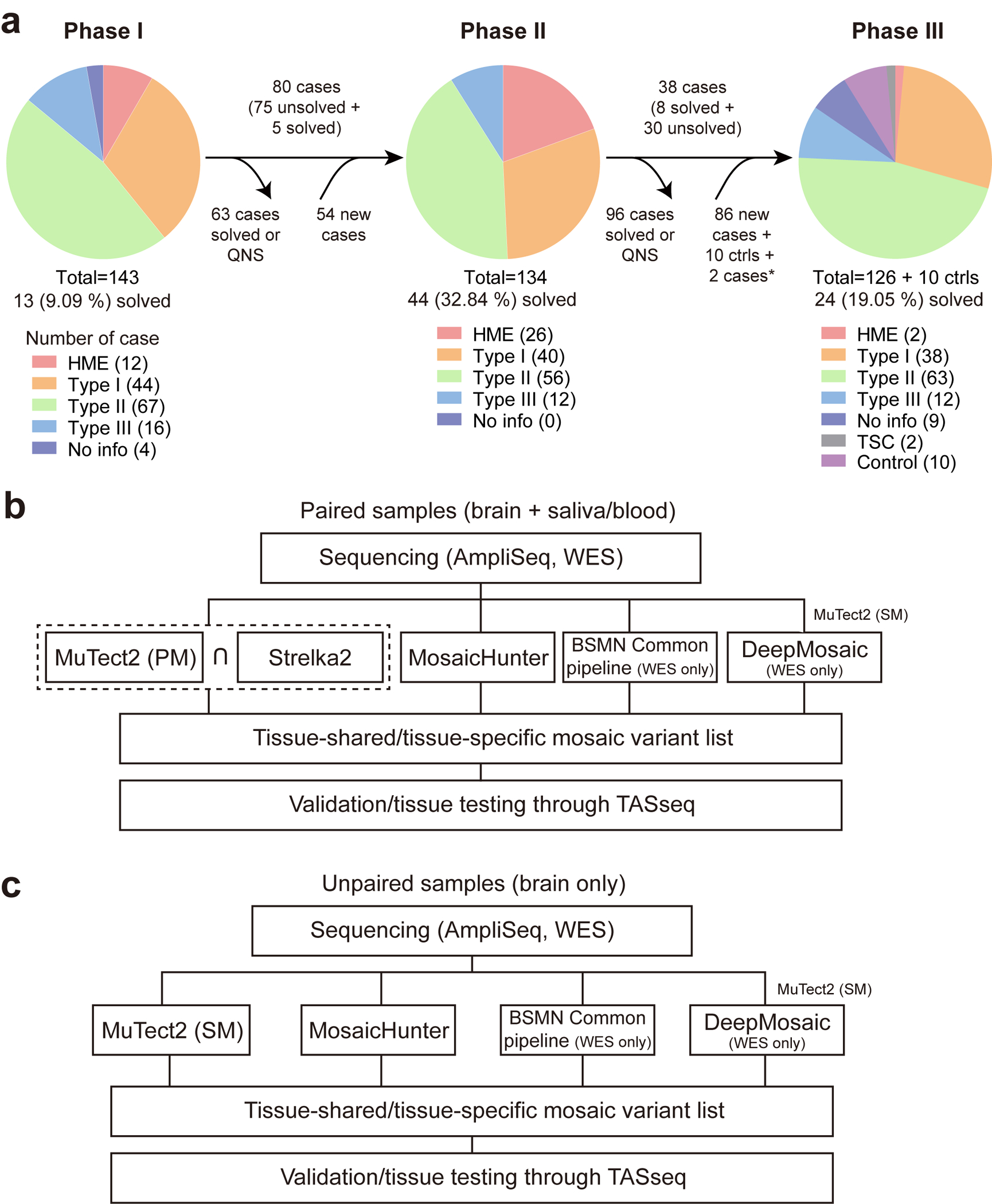
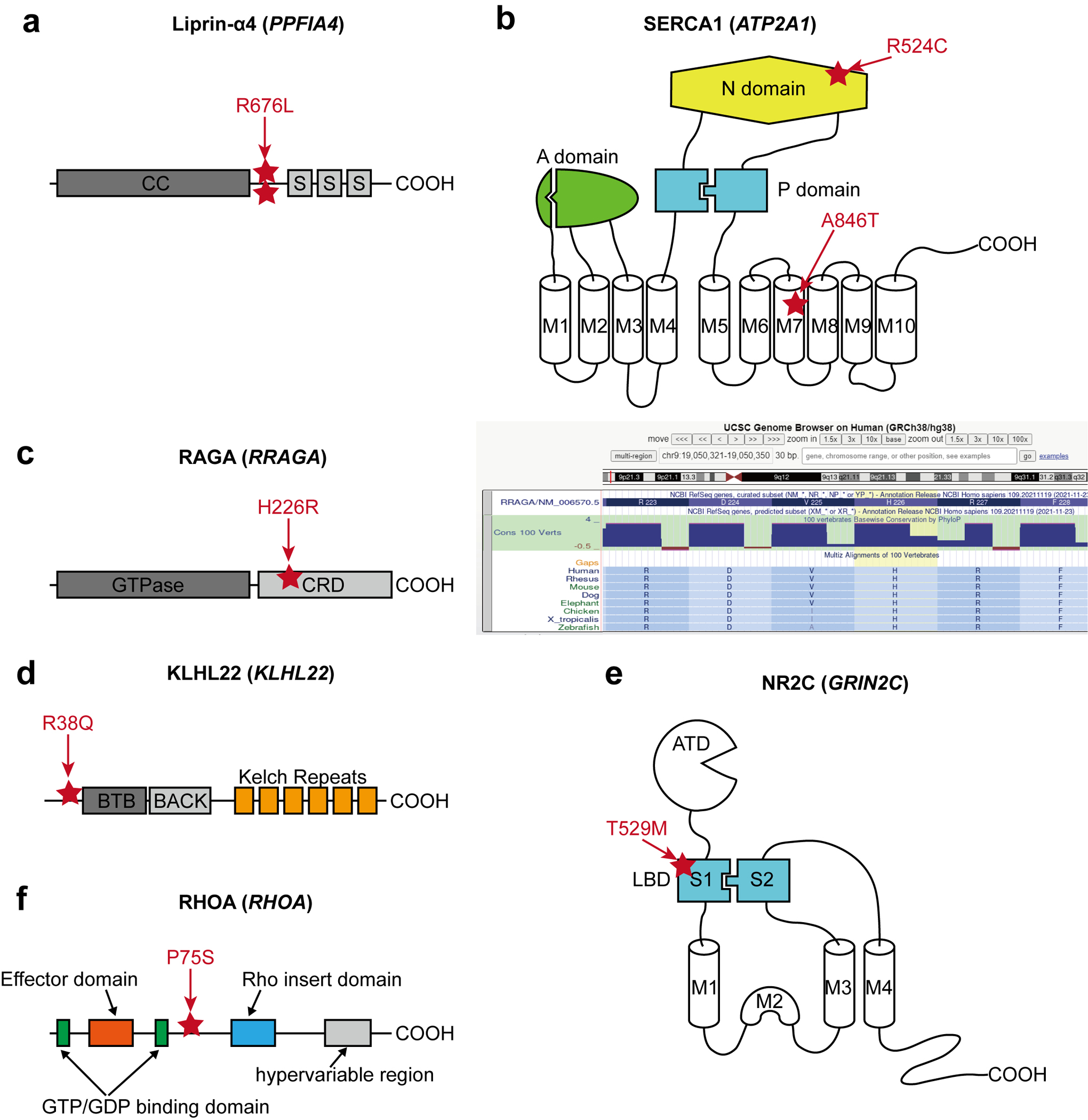
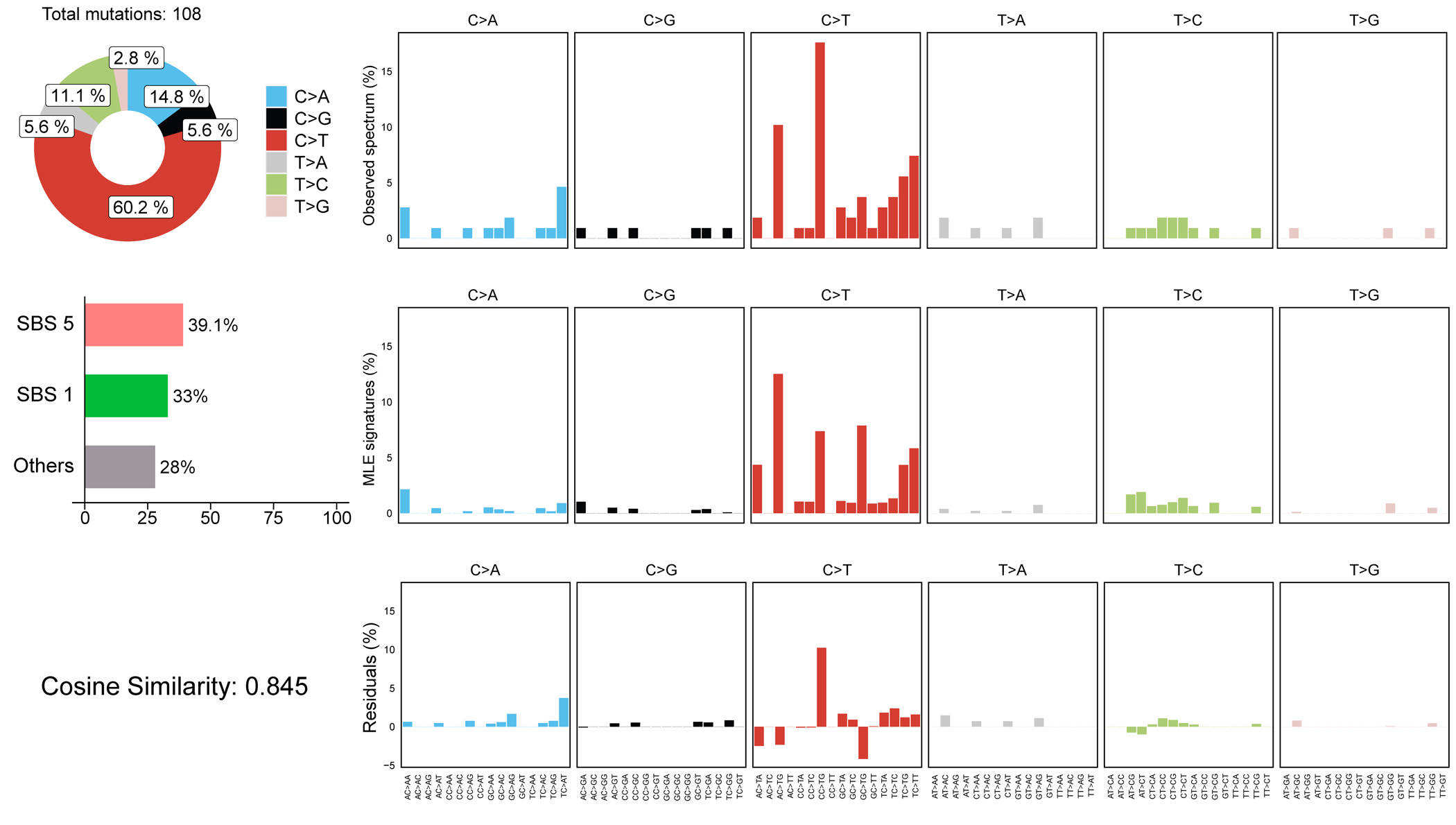
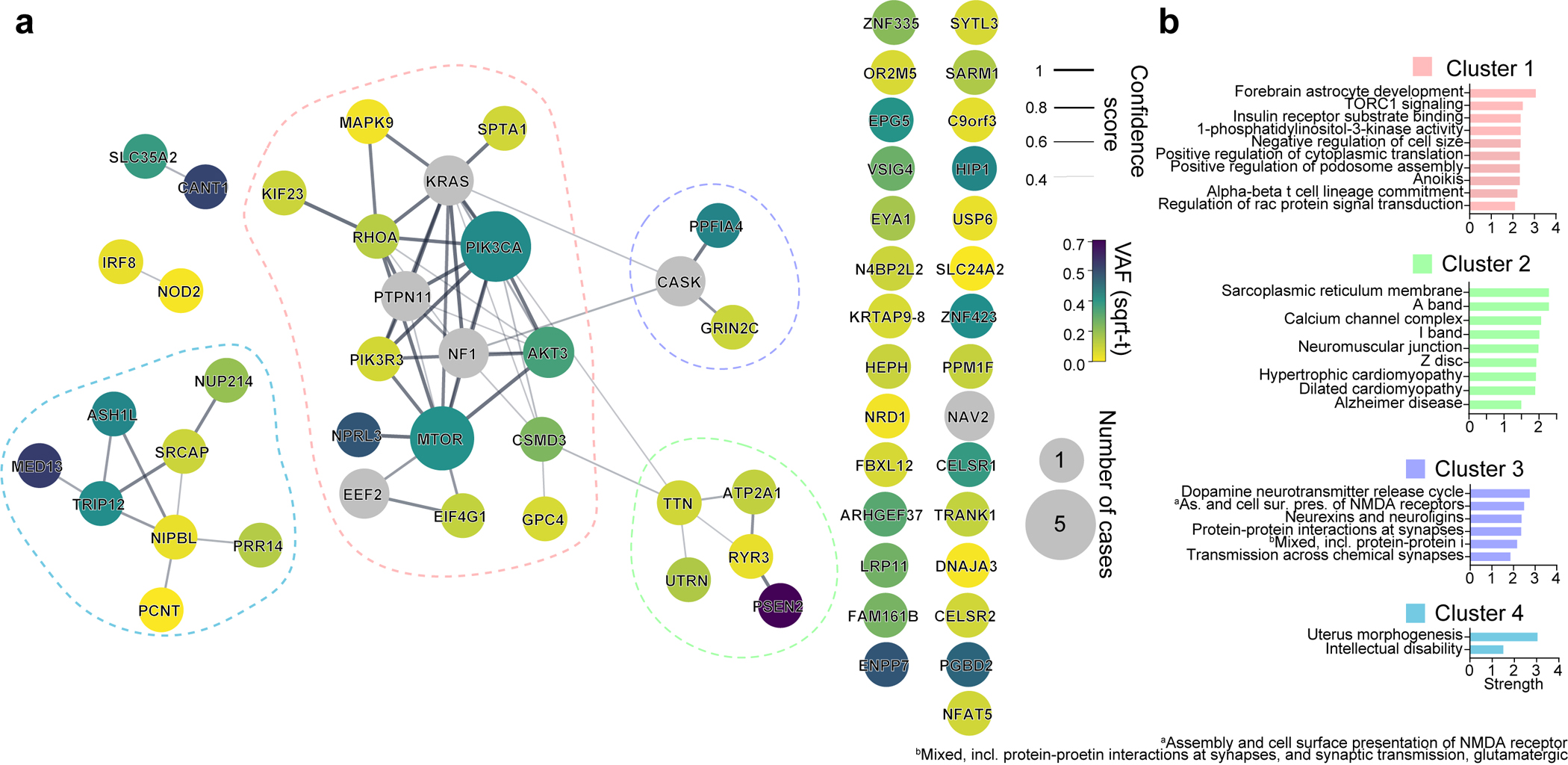
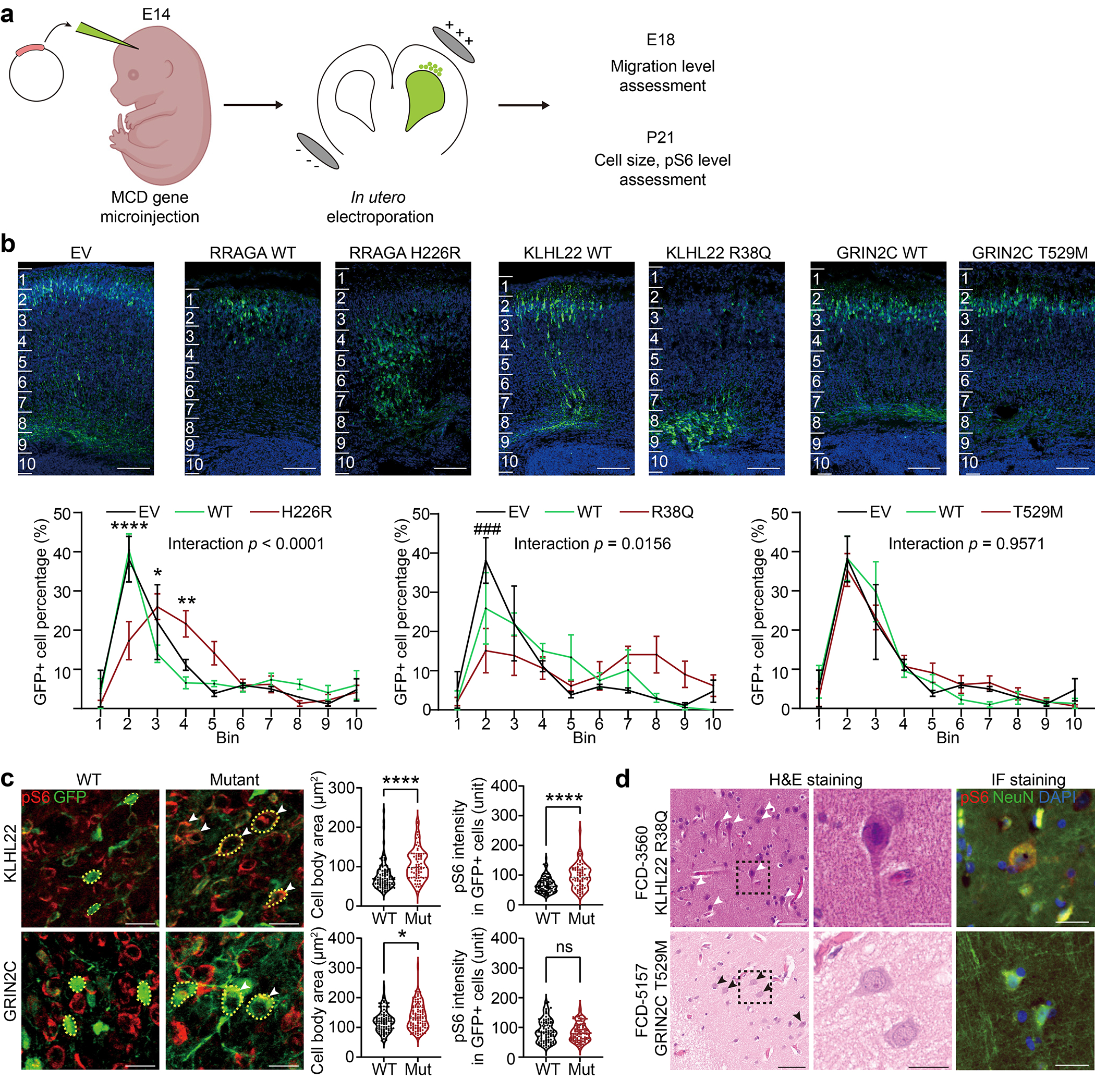
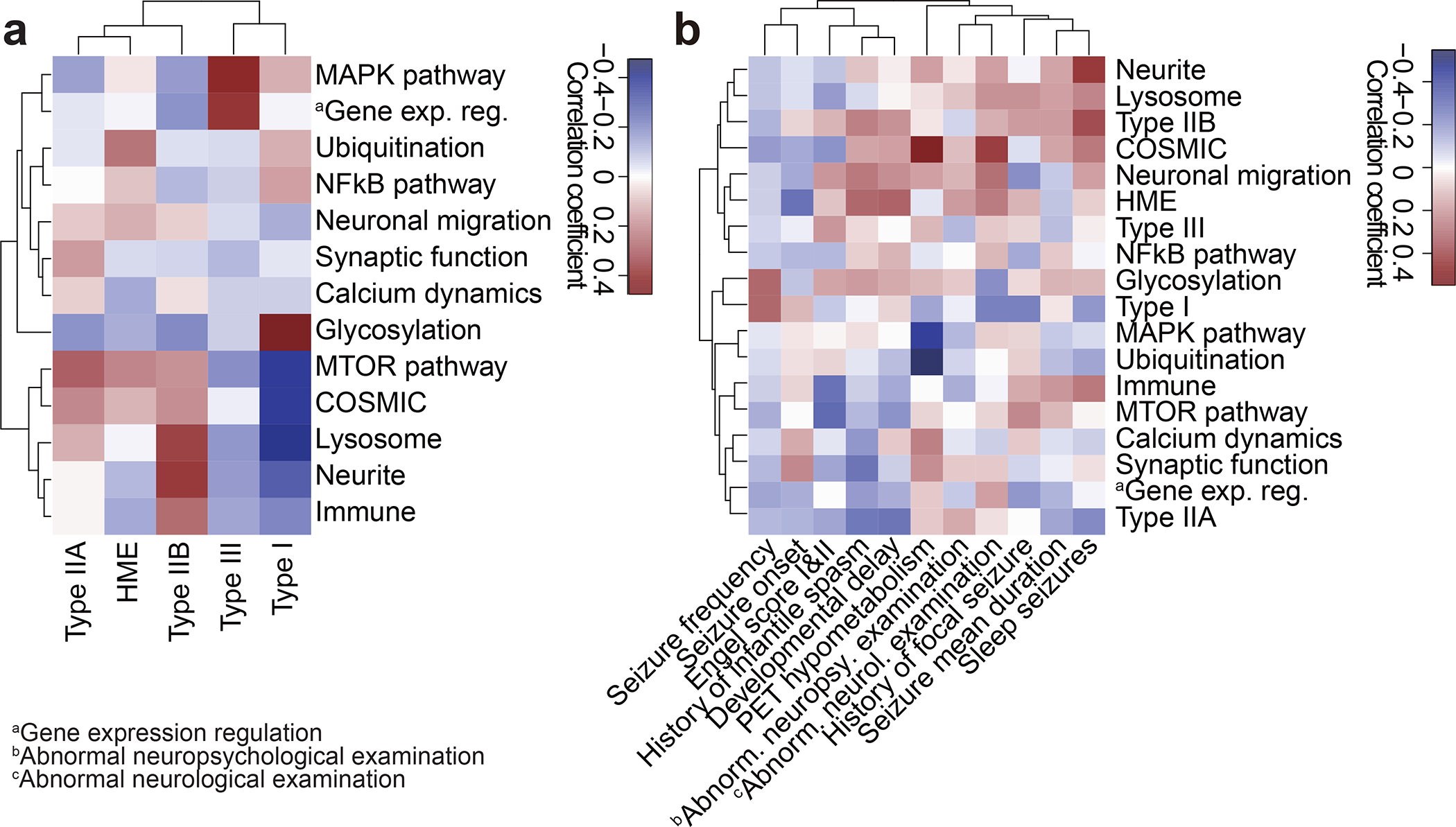
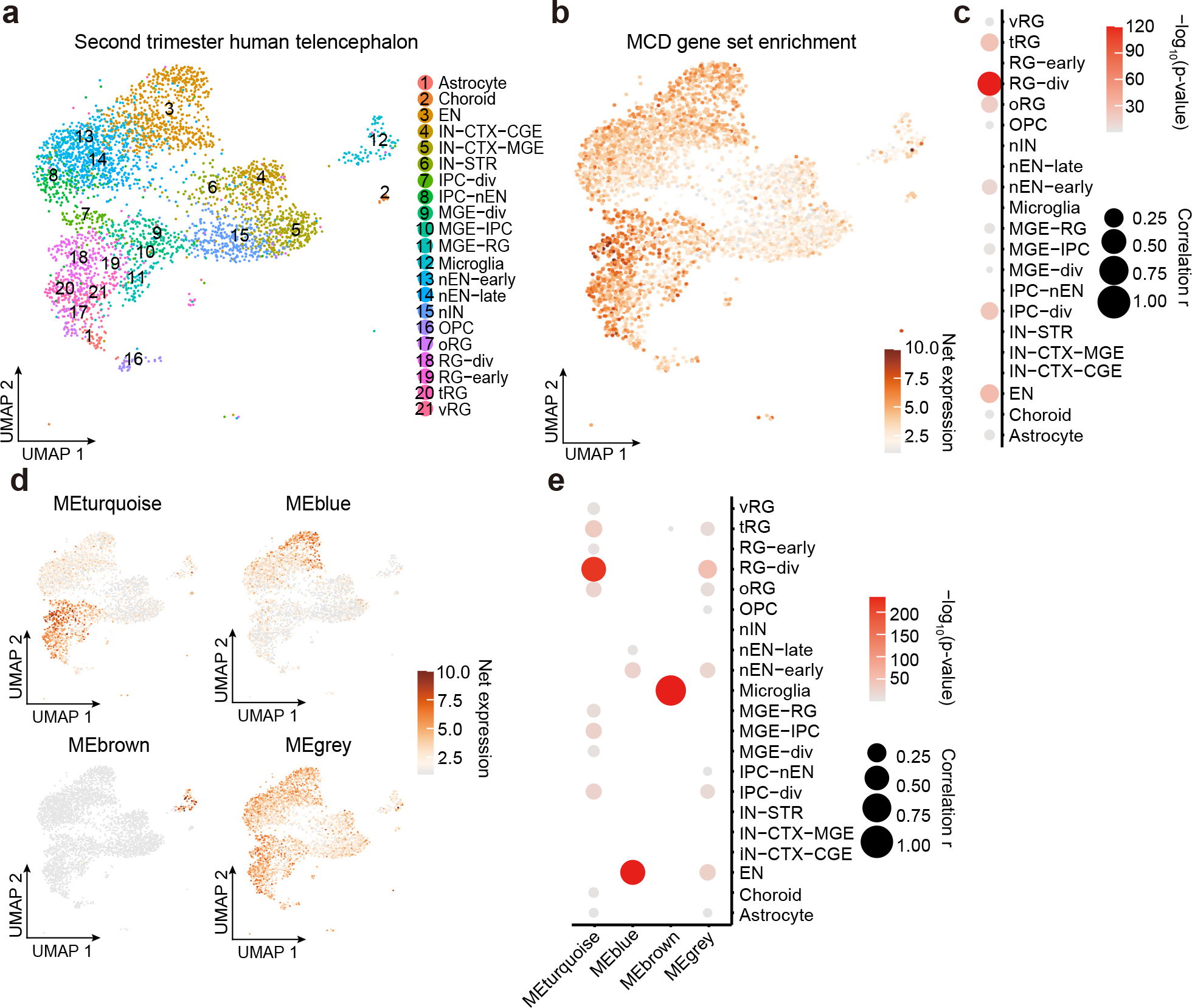
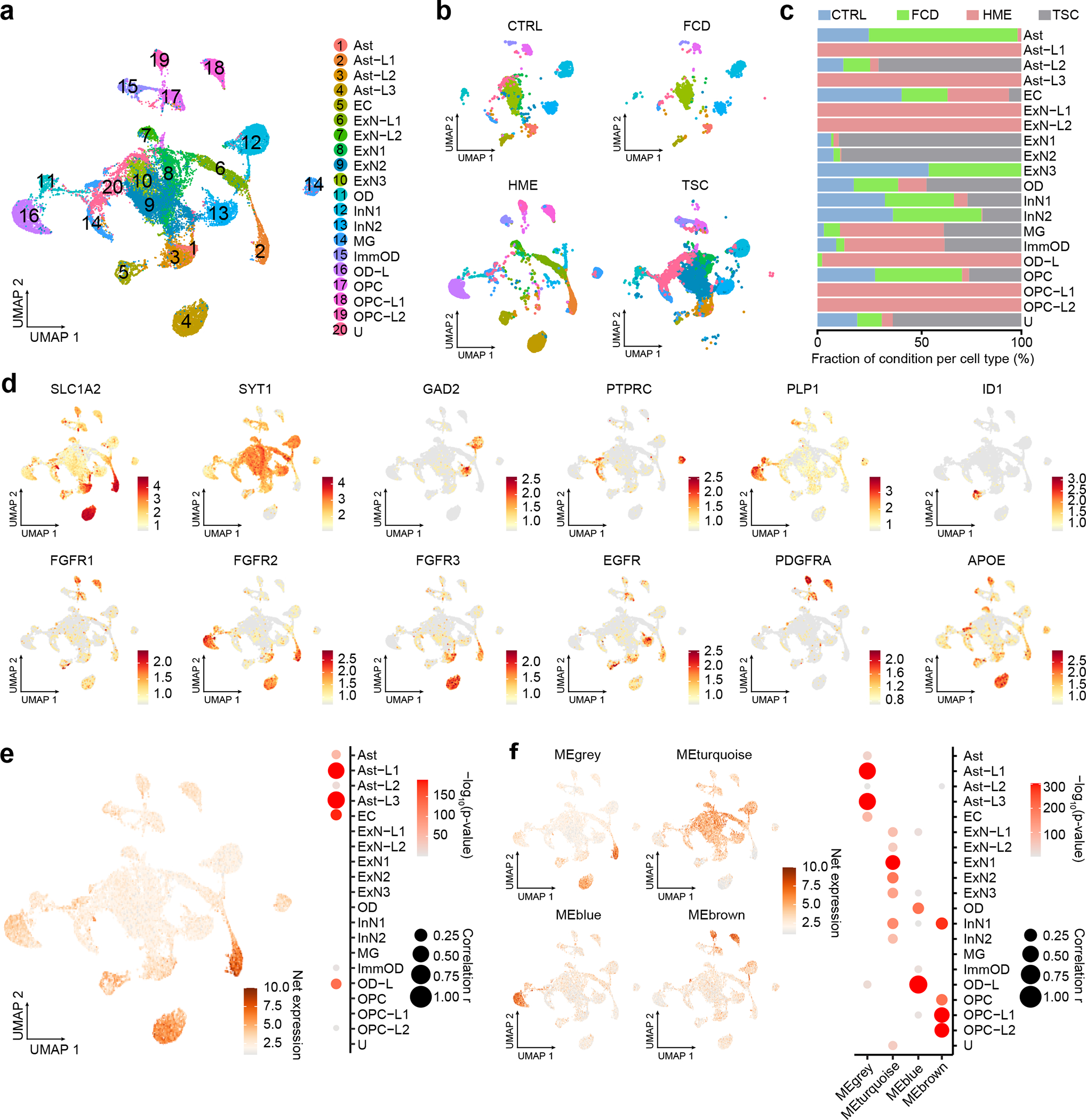
References
-
- Krsek P et al. Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 63, 758–69 (2008). - PubMed
Method-only references
-
- Kim S et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594 (2018). - PubMed
-
- Benjamin D et al. Calling Somatic SNVs and Indels with Mutect2. bioRxiv (2019).
Publication types
MeSH terms
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- R01 MH124890/MH/NIMH NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- U54 HG006504/HG/NHGRI NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- T32 GM007544/GM/NIGMS NIH HHS/United States
- U24 NS120854/NS/NINDS NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U01 MH108898/MH/NIMH NIH HHS/United States
- P50 HD103525/HD/NICHD NIH HHS/United States
- R21 AG070462/AG/NIA NIH HHS/United States
- R01 NS083823/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
