

OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases
- PMID: 36635485
- PMCID: PMC9837150
- DOI: 10.1038/s42003-022-04303-x
OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases
Abstract
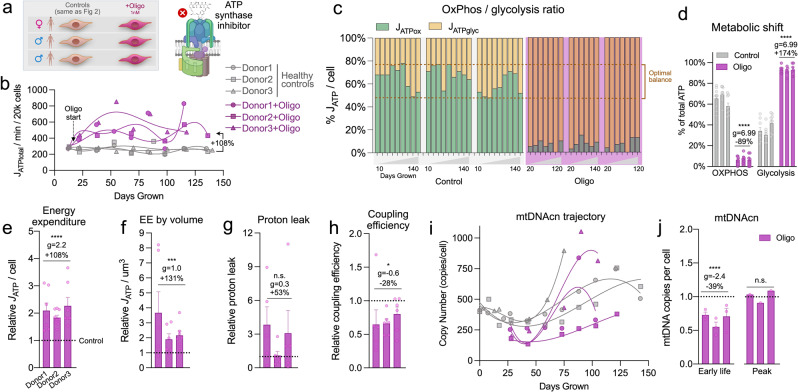
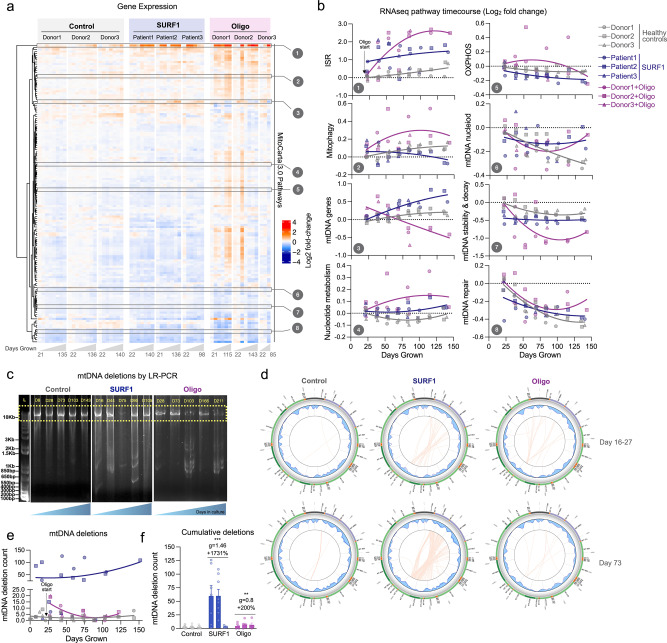
Patients with primary mitochondrial oxidative phosphorylation (OxPhos) defects present with fatigue and multi-system disorders, are often lean, and die prematurely, but the mechanistic basis for this clinical picture remains unclear. By integrating data from 17 cohorts of patients with mitochondrial diseases (n = 690) we find evidence that these disorders increase resting energy expenditure, a state termed hypermetabolism. We examine this phenomenon longitudinally in patient-derived fibroblasts from multiple donors. Genetically or pharmacologically disrupting OxPhos approximately doubles cellular energy expenditure. This cell-autonomous state of hypermetabolism occurs despite near-normal OxPhos coupling efficiency, excluding uncoupling as a general mechanism. Instead, hypermetabolism is associated with mitochondrial DNA instability, activation of the integrated stress response (ISR), and increased extracellular secretion of age-related cytokines and metabokines including GDF15. In parallel, OxPhos defects accelerate telomere erosion and epigenetic aging per cell division, consistent with evidence that excess energy expenditure accelerates biological aging. To explore potential mechanisms for these effects, we generate a longitudinal RNASeq and DNA methylation resource dataset, which reveals conserved, energetically demanding, genome-wide recalibrations. Taken together, these findings highlight the need to understand how OxPhos defects influence the energetic cost of living, and the link between hypermetabolism and aging in cells and patients with mitochondrial diseases.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
