

3D-equivariant graph neural networks for protein model quality assessment
- PMID: 36637199
- PMCID: PMC10089647
- DOI: 10.1093/bioinformatics/btad030
3D-equivariant graph neural networks for protein model quality assessment
Abstract
Motivation: Quality assessment (QA) of predicted protein tertiary structure models plays an important role in ranking and using them. With the recent development of deep learning end-to-end protein structure prediction techniques for generating highly confident tertiary structures for most proteins, it is important to explore corresponding QA strategies to evaluate and select the structural models predicted by them since these models have better quality and different properties than the models predicted by traditional tertiary structure prediction methods.
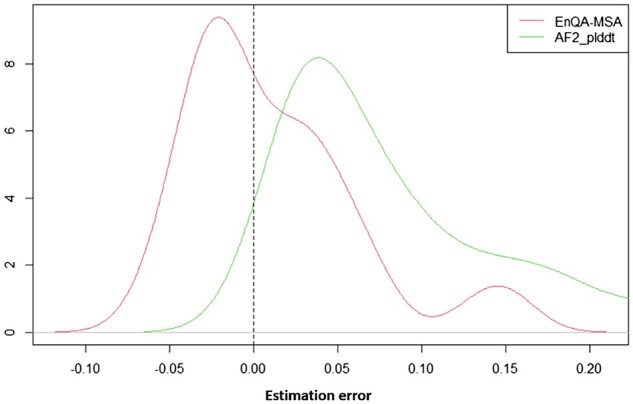
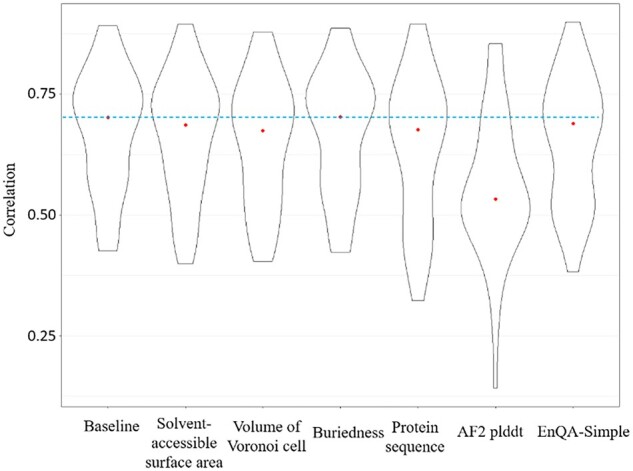
Results: We develop EnQA, a novel graph-based 3D-equivariant neural network method that is equivariant to rotation and translation of 3D objects to estimate the accuracy of protein structural models by leveraging the structural features acquired from the state-of-the-art tertiary structure prediction method-AlphaFold2. We train and test the method on both traditional model datasets (e.g. the datasets of the Critical Assessment of Techniques for Protein Structure Prediction) and a new dataset of high-quality structural models predicted only by AlphaFold2 for the proteins whose experimental structures were released recently. Our approach achieves state-of-the-art performance on protein structural models predicted by both traditional protein structure prediction methods and the latest end-to-end deep learning method-AlphaFold2. It performs even better than the model QA scores provided by AlphaFold2 itself. The results illustrate that the 3D-equivariant graph neural network is a promising approach to the evaluation of protein structural models. Integrating AlphaFold2 features with other complementary sequence and structural features is important for improving protein model QA.
Availability and implementation: The source code is available at https://github.com/BioinfoMachineLearning/EnQA.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2023. Published by Oxford University Press.
Figures
References
-
- Arnold K. et al. (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics, 22, 195–201. - PubMed
