

GALA: a computational framework for de novo chromosome-by-chromosome assembly with long reads
- PMID: 36639368
- PMCID: PMC9839709
- DOI: 10.1038/s41467-022-35670-y
GALA: a computational framework for de novo chromosome-by-chromosome assembly with long reads
Abstract
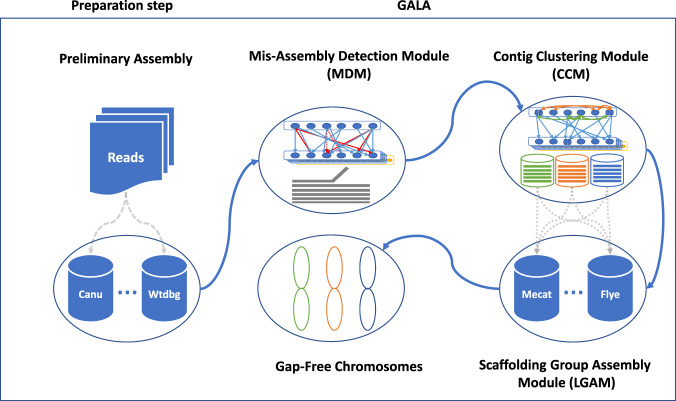
High-quality genome assembly has wide applications in genetics and medical studies. However, it is still very challenging to achieve gap-free chromosome-scale assemblies using current workflows for long-read platforms. Here we report on GALA (Gap-free long-read Assembly tool), a computational framework for chromosome-based sequencing data separation and de novo assembly implemented through a multi-layer graph that identifies discordances within preliminary assemblies and partitions the data into chromosome-scale scaffolding groups. The subsequent independent assembly of each scaffolding group generates a gap-free assembly likely free from the mis-assembly errors which usually hamper existing workflows. This flexible framework also allows us to integrate data from various technologies, such as Hi-C, genetic maps, and even motif analyses to generate gap-free chromosome-scale assemblies. As a proof of principle we de novo assemble the C. elegans genome using combined PacBio and Nanopore sequencing data and a rice cultivar genome using Nanopore sequencing data from publicly available datasets. We also demonstrate the proposed method's applicability with a gap-free assembly of the human genome using PacBio high-fidelity (HiFi) long reads. Thus, our method enables straightforward assembly of genomes with multiple data sources and overcomes barriers that at present restrict the application of de novo genome assembly technology.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
