

Transformer for one stop interpretable cell type annotation
- PMID: 36641532
- PMCID: PMC9840170
- DOI: 10.1038/s41467-023-35923-4
Transformer for one stop interpretable cell type annotation
Abstract
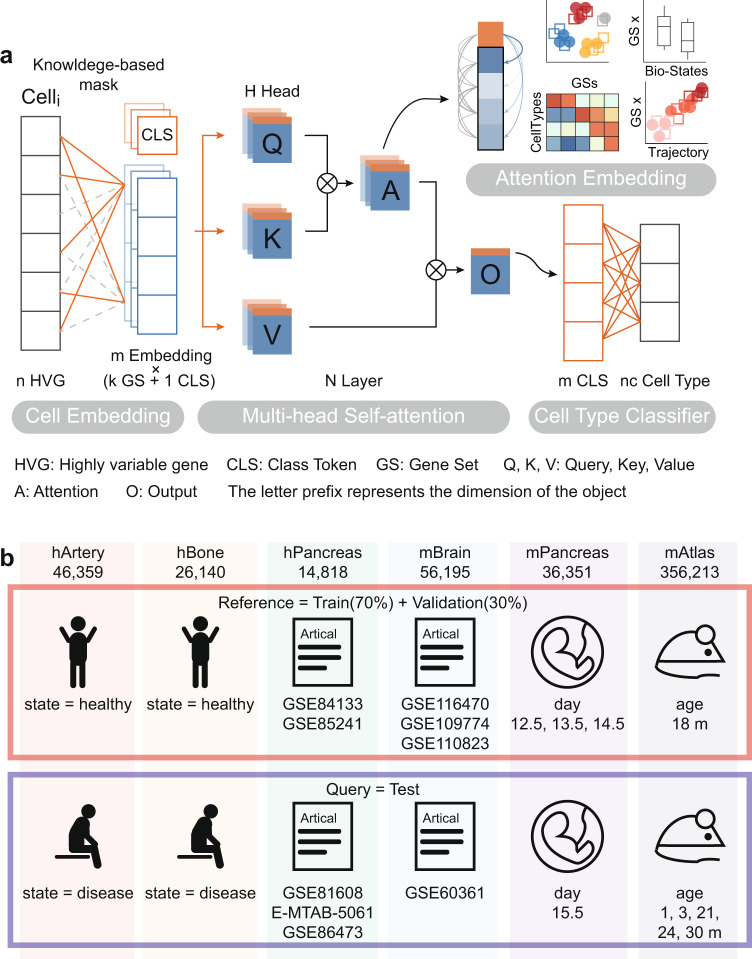
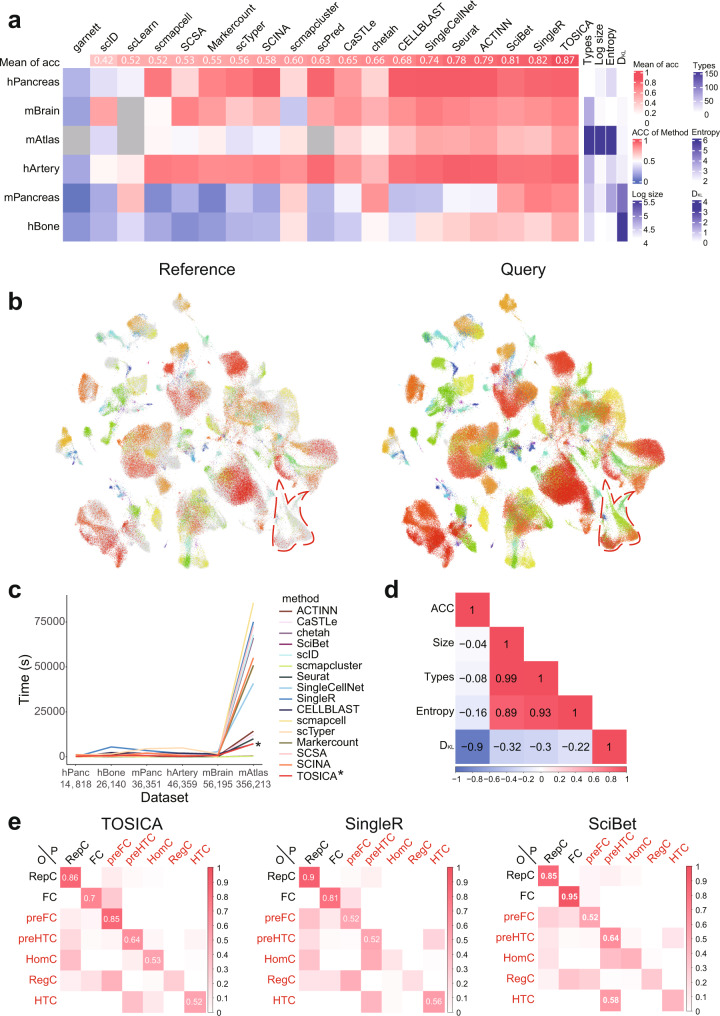
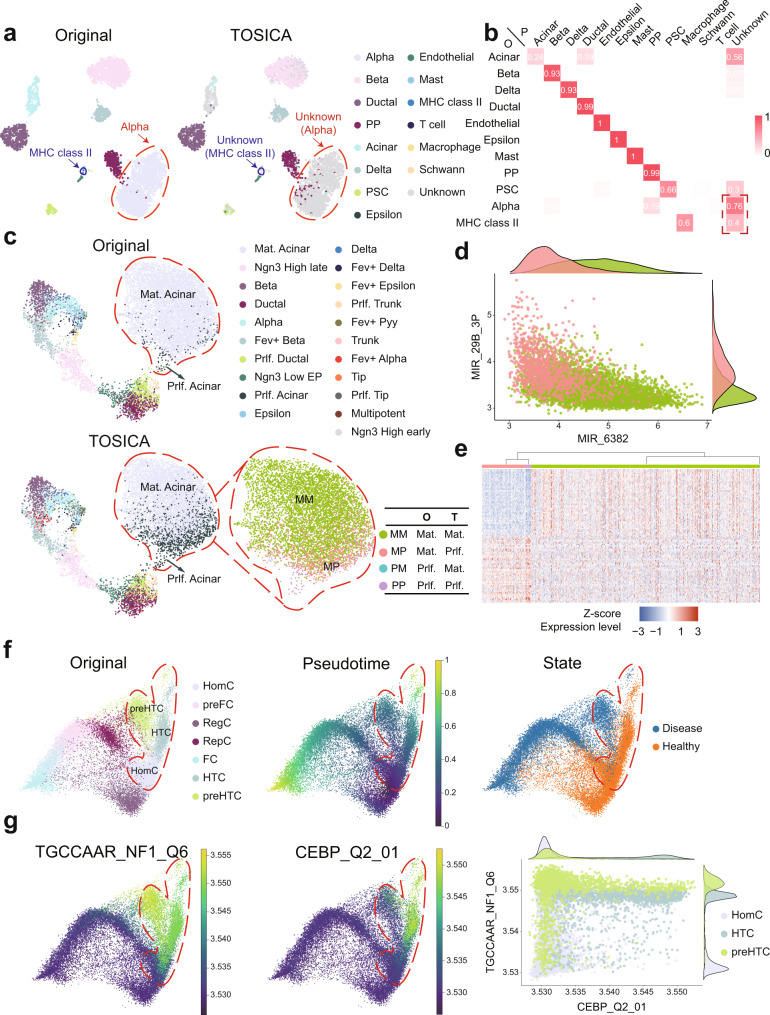
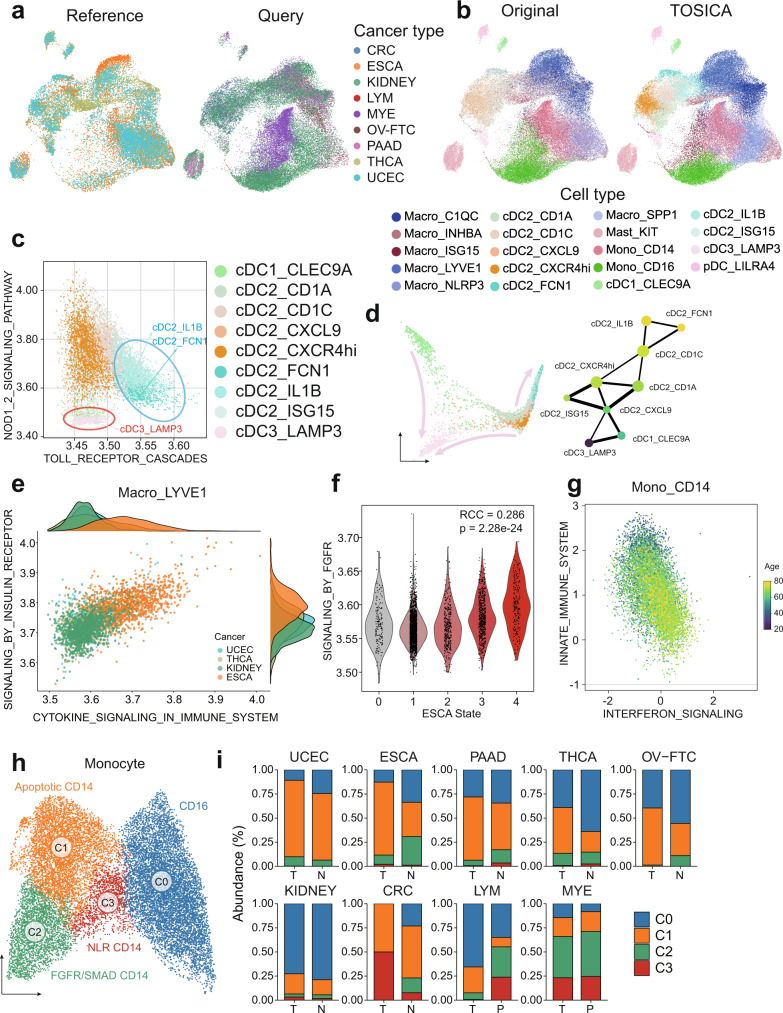
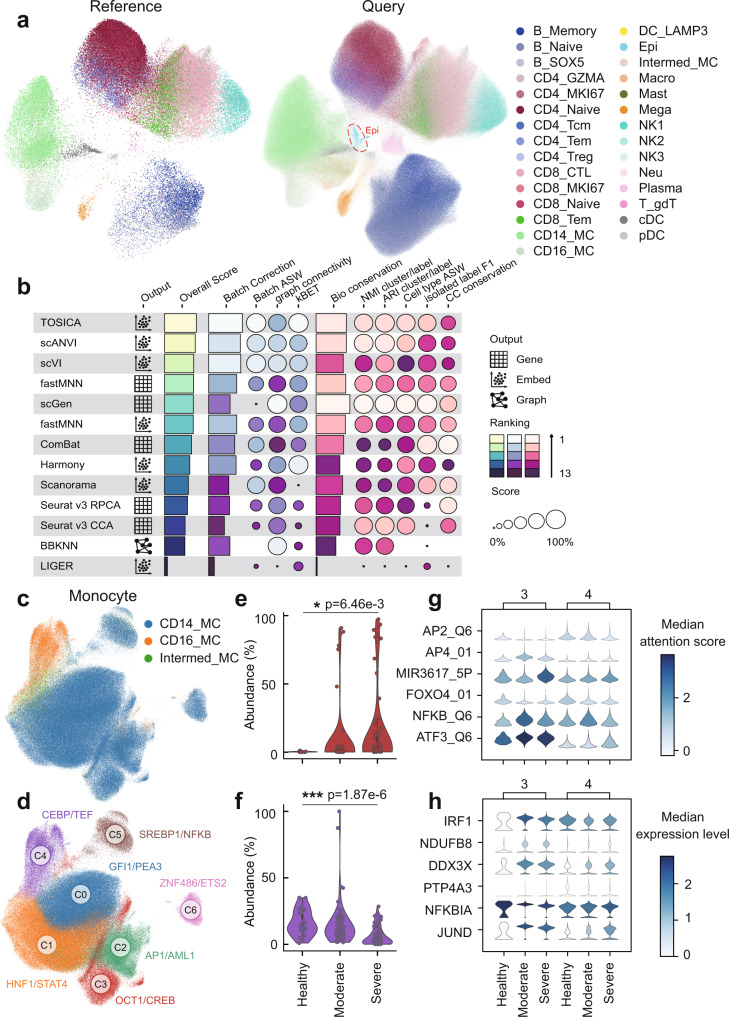
Consistent annotation transfer from reference dataset to query dataset is fundamental to the development and reproducibility of single-cell research. Compared with traditional annotation methods, deep learning based methods are faster and more automated. A series of useful single cell analysis tools based on autoencoder architecture have been developed but these struggle to strike a balance between depth and interpretability. Here, we present TOSICA, a multi-head self-attention deep learning model based on Transformer that enables interpretable cell type annotation using biologically understandable entities, such as pathways or regulons. We show that TOSICA achieves fast and accurate one-stop annotation and batch-insensitive integration while providing biologically interpretable insights for understanding cellular behavior during development and disease progressions. We demonstrate TOSICA's advantages by applying it to scRNA-seq data of tumor-infiltrating immune cells, and CD14+ monocytes in COVID-19 to reveal rare cell types, heterogeneity and dynamic trajectories associated with disease progression and severity.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
