

MCU Upregulation Overactivates Mitophagy by Promoting VDAC1 Dimerization and Ubiquitination in the Hepatotoxicity of Cadmium
- PMID: 36642847
- PMCID: PMC9982555
- DOI: 10.1002/advs.202203869
MCU Upregulation Overactivates Mitophagy by Promoting VDAC1 Dimerization and Ubiquitination in the Hepatotoxicity of Cadmium
Abstract
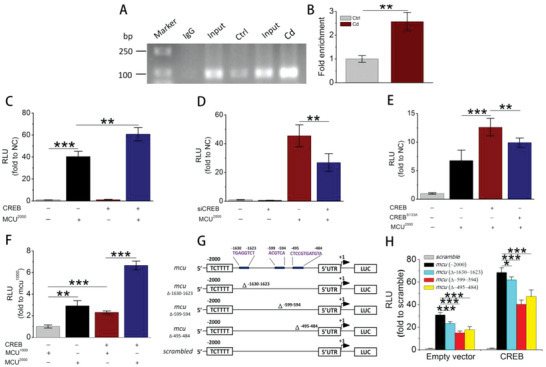
Cadmium (Cd) is a high-risk pathogenic toxin for hepatic diseases. Excessive mitophagy is a hallmark in Cd-induced hepatotoxicity. However, the underlying mechanism remains obscure. Mitochondrial calcium uniporter (MCU) is a key regulator for mitochondrial and cellular homeostasis. Here, Cd exposure upregulated MCU expression and increased mitochondrial Ca2+ uptake are found. MCU inhibition through siRNA or by Ru360 significantly attenuates Cd-induced excessive mitophagy, thereby rescues mitochondrial dysfunction and increases hepatocyte viability. Heterozygous MCU knockout mice exhibit improved liver function, ameliorated pathological damage, less mitochondrial fragmentation, and mitophagy after Cd exposure. Mechanistically, Cd upregulates MCU expression through phosphorylation activation of cAMP-response element binding protein at Ser133(CREBS133 ) and subsequent binding of MCU promoter at the TGAGGTCT, ACGTCA, and CTCCGTGATGTA regions, leading to increased MCU gene transcription. The upregulated MCU intensively interacts with voltage-dependent anion-selective channel protein 1 (VDAC1), enhances its dimerization and ubiquitination, resulting in excessive mitophagy. This study reveals a novel mechanism, through which Cd upregulates MCU to enhance mitophagy and hepatotoxicity.
Keywords: cadmium; hepatotoxicity; mitochondrial calcium uniporter; mitophagy; voltage-dependent anion-selective channel protein 1.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
-
- a) Flannery B. M., Schaefer H. R., Middleton K. B., Regul. Toxicol. Pharmacol. 2022, 131, 105155; - PubMed
- b) Satarug S., Baker J. R., Urbenjapol S., Haswell‐Elkins M., Reilly P. E., Williams D. J., Moore M. R., Toxicol. Lett. 2003, 137, 65; - PubMed
- c) Genchi G., Sinicropi M. S., Lauria G., Carocci A., Catalano A., Int. J. Environ. Res. Public Health 2020, 17, 3782. - PubMed
-
- a) Hong D., Min J. Y., Min K. B., J. Prev. Med. Public Health 2021, 54, 471; - PMC - PubMed
- b) Park E., Kim J., Kim B., Park E. Y., Chemosphere 2021, 266, 128947; - PubMed
- c) Han S., Sung G. H., Lee S., Han K. J., Han H. J., Medicine 2022, 101, 28559; - PubMed
- d) Li Y., Chen C., Lu L., Guo W., VanWagner L. B., Shikany J. M., Zhang S., Kahe K., Dig. Dis. Sci. 2022, 67, 689; - PMC - PubMed
- e) Satarug S., Curr. Drug Metab. 2012, 13, 257; - PubMed
- f) Hyder O., Chung M., Cosgrove D., Herman J. M., Li Z., Firoozmand A., Gurakar A., Koteish A., Pawlik T. M., J. Gastrointest. Surg. 2013, 17, 1265. - PMC - PubMed
-
- a) Men H., Young J. L., Zhou W., Zhang H., Wang X., Xu J., Lin Q., Tan Y., Zheng Y., Cai L., Oxid. Med. Cell. Longevity 2021, 2021, 1427787; - PMC - PubMed
- b) Li X., Li H., Cai D., Li P., Jin J., Jiang X., Li Z., Tian L., Chen G., Sun J., Bai W., Food Chem. Toxicol. 2021, 148, 111944; - PubMed
- c) Alshammari G. M., Al‐Qahtani W. H., AlFaris N. A., Alzahrani N. S., Alkhateeb M. A., Yahya M. A., BioFactors 2021, 47, 489. - PubMed
-
- a) Xu S., Pi H., Chen Y., Zhang N., Guo P., Lu Y., He M., Xie J., Zhong M., Zhang Y., Yu Z., Zhou Z., Cell Death Dis. 2013, 4, 540; - PMC - PubMed
- b) Pi H., Xu S., Zhang L., Guo P., Li Y., Xie J., Tian L., He M., Lu Y., Li M., Zhang Y., Zhong M., Xiang Y., Deng L., Zhou Z., Yu Z., Autophagy 2013, 9, 1780. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 82073523/National Natural Science Foundation of China
- cstc2020jcyj-jqx0027/Chongqing Science Foundation for Distinguished Young Scholars
- CQYC201905029/Chongqing Youth Talent Project
- 2019ZDXM019/Chongqing Science and Health Joint Medical Research Key Project
- CSTB2022NSCQ-MSX1142/Chongqing Natural Science Foundation
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous