

Collective Variables for Crystallization Simulations-from Early Developments to Recent Advances
- PMID: 36643553
- PMCID: PMC9835087
- DOI: 10.1021/acsomega.2c06310
Collective Variables for Crystallization Simulations-from Early Developments to Recent Advances
Abstract
Crystallization is an important physicochemical process which has relevance in material science, biology, and the environment. Decades of experimental and theoretical efforts have been made to understand this fundamental symmetry-breaking transition. While experiments provide equilibrium structures and shapes of crystals, they are limited to unraveling how molecules aggregate to form crystal nuclei that subsequently transform into bulk crystals. Computer simulations, mainly molecular dynamics (MD), can provide such microscopic details during the early stage of a crystallization event. Crystallization is a rare event that takes place in time scales much longer than a typical equilibrium MD simulation can sample. This inadequate sampling of the MD method can be easily circumvented by the use of enhanced sampling (ES) simulations. In most of the ES methods, the fluctuations of a system's slow degrees of freedom, called collective variables (CVs), are enhanced by applying a bias potential. This transforms the system from one state to the other within a short time scale. The most crucial part of such CV-based ES methods is to find suitable CVs, which often needs intuition and several trial-and-error optimization steps. Over the years, a plethora of CVs has been developed and applied in the study of crystallization. In this review, we provide a brief overview of CVs that have been developed and used in ES simulations to study crystallization from melt or solution. These CVs can be categorized mainly into four types: (i) spherical particle-based, (ii) molecular template-based, (iii) physical property-based, and (iv) CVs obtained from dimensionality reduction techniques. We present the context-based evolution of CVs, discuss the current challenges, and propose future directions to further develop effective CVs for the study of crystallization of complex systems.
© 2022 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures




















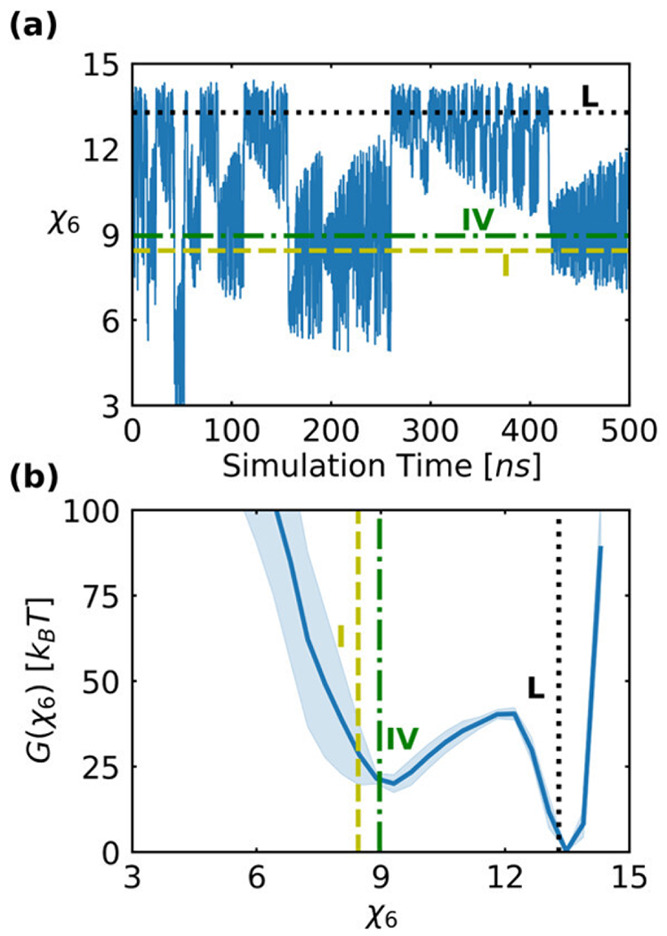

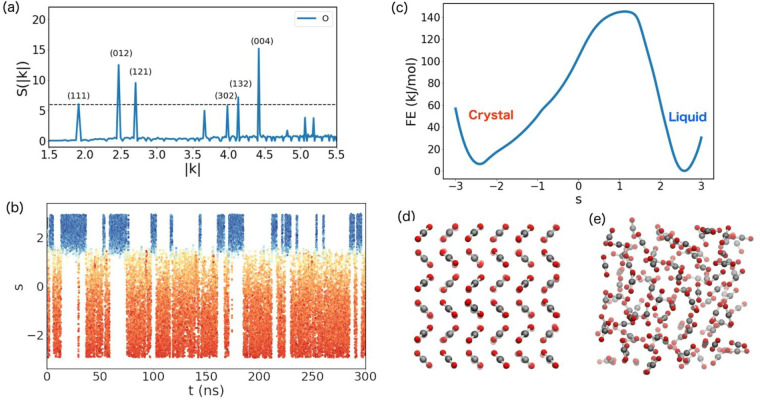

References
-
- Torrie G. M.; Valleau J. P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1997, 23, 187–199. 10.1016/0021-9991(77)90121-8. - DOI
-
- Nakajima N.; Nakamura H.; Kidera A. Multicanonical ensemble generated by molecular dynamics simulation for enhanced conformational sampling of peptides. J. Phys. Chem. B 1997, 101, 817–824. 10.1021/jp962142e. - DOI
-
- Sugita Y.; Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chemical physics letters 1999, 314, 141–151. 10.1016/S0009-2614(99)01123-9. - DOI
-
- Sorensen M. R.; Voter A. F. Temperature-accelerated dynamics for simulation of infrequent events. J. Chem. Phys. 2000, 112, 9599–9606. 10.1063/1.481576. - DOI
Publication types
LinkOut - more resources
Full Text Sources
