

LncRNA BC promotes lung adenocarcinoma progression by modulating IMPAD1 alternative splicing
- PMID: 36650118
- PMCID: PMC9845120
- DOI: 10.1002/ctm2.1129
LncRNA BC promotes lung adenocarcinoma progression by modulating IMPAD1 alternative splicing
Abstract
Background: The therapeutic value of targeted therapies in patients with lung cancer is reduced when tumours acquire secondary resistance after an initial period of successful treatment. However, the molecular events behind the resistance to targeted therapies in lung cancer remain largely unknown.
Aims: To discover the important role and mechanism of lncRNA BC in promoting tumor metastasis and influencing clinical prognosis of LUAD.
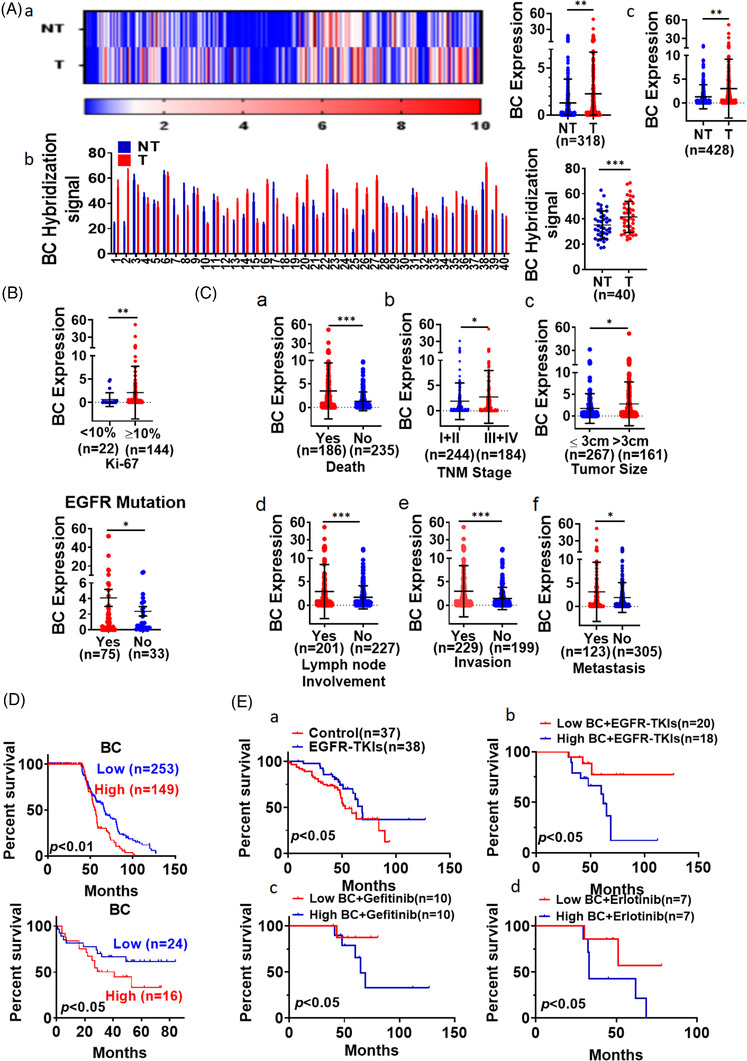
Materials & methods: Microarrays were used to screen a comprehensive set of lncRNAs with differential expression profiles in lung cancer cells. The functional role and mechanism of lncRNA were further investigated by gain- and loss-of-function assays. RNA pull-down, protein assays, and mass spectrometry were used to identify proteins that interacted with lncRNA. TaqMan PCR was used to measure lncRNA in lung adenocarcinoma and adjacent nontumor tissues from 428 patients. The clinical significance of lncRNA identified was statistically confirmed in this cohort of patients.
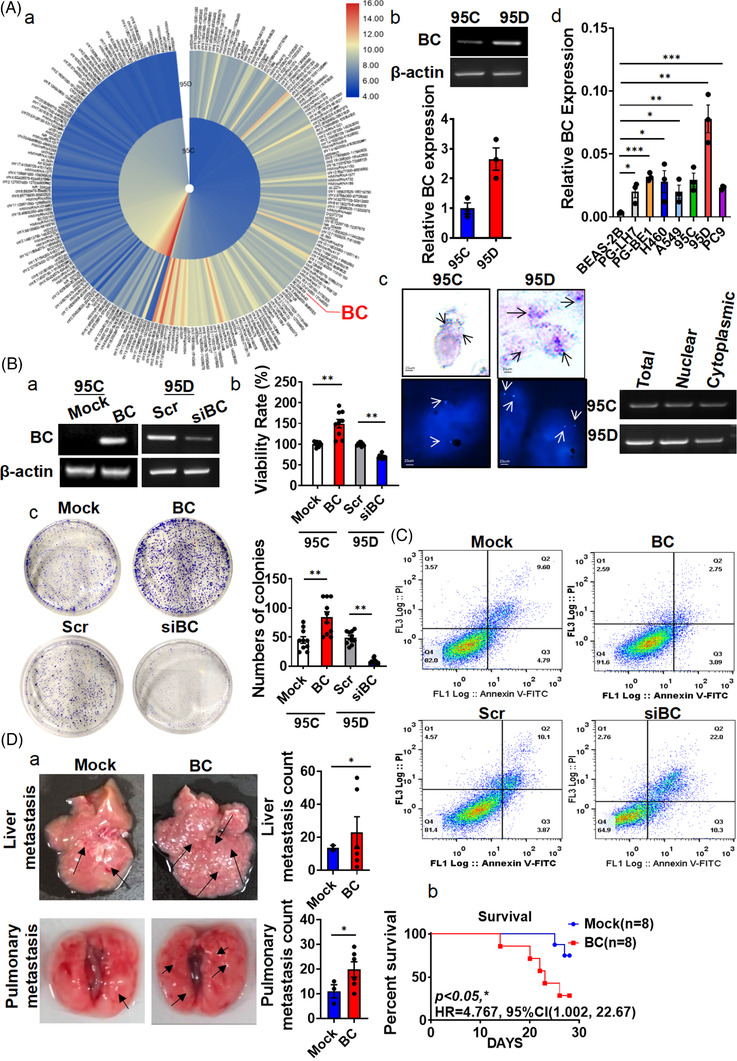
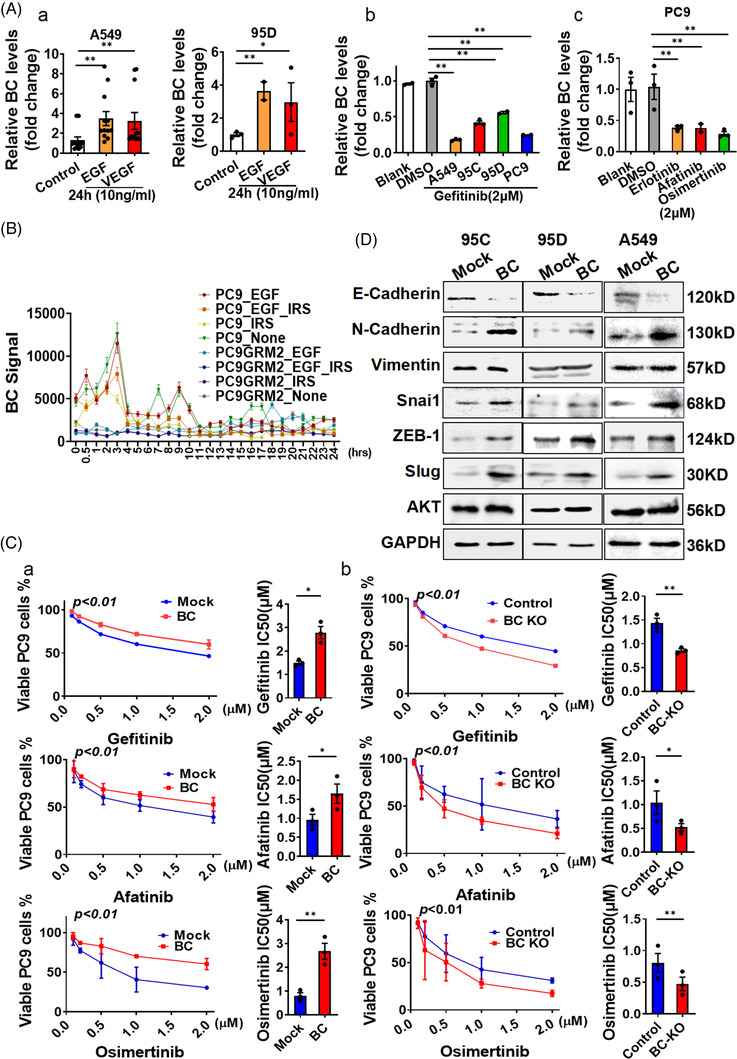
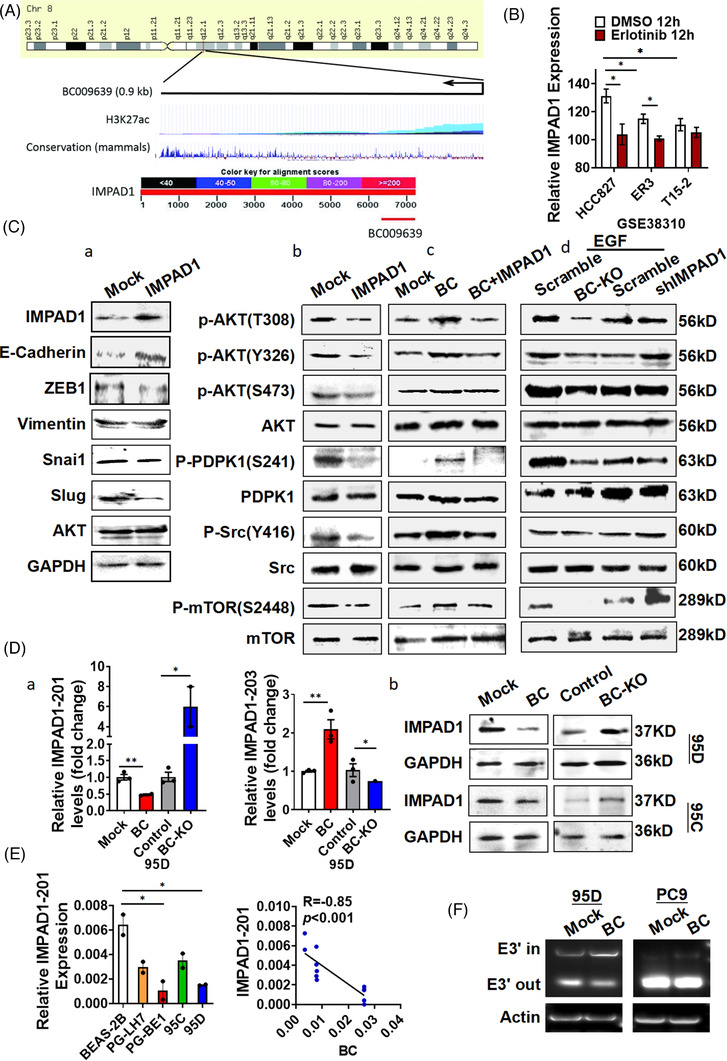
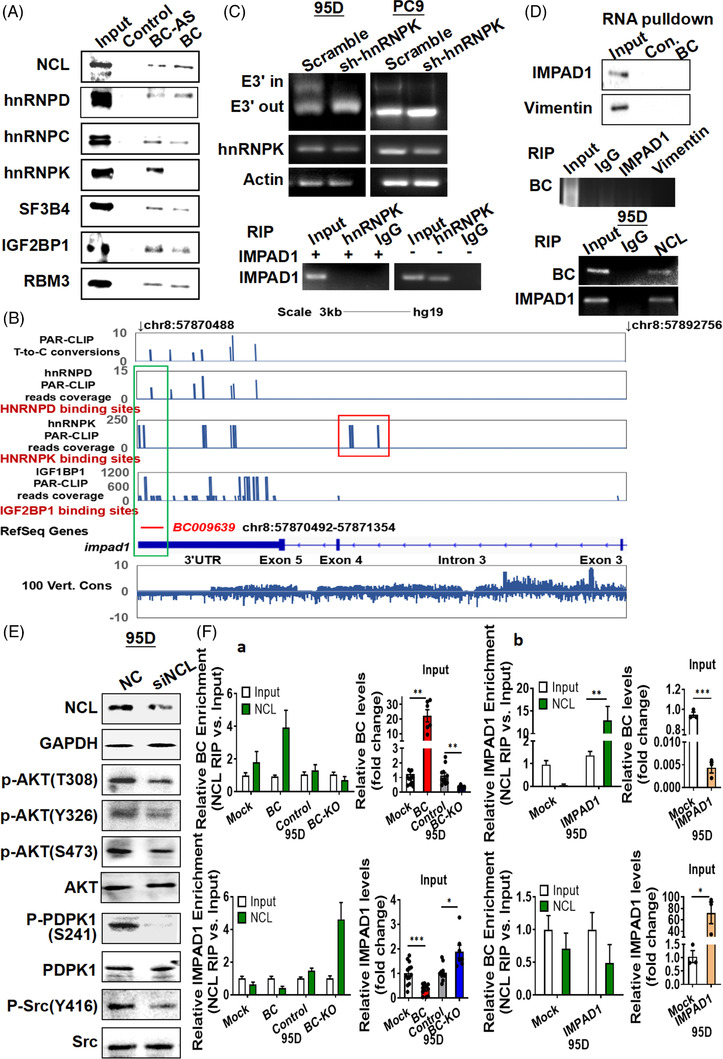
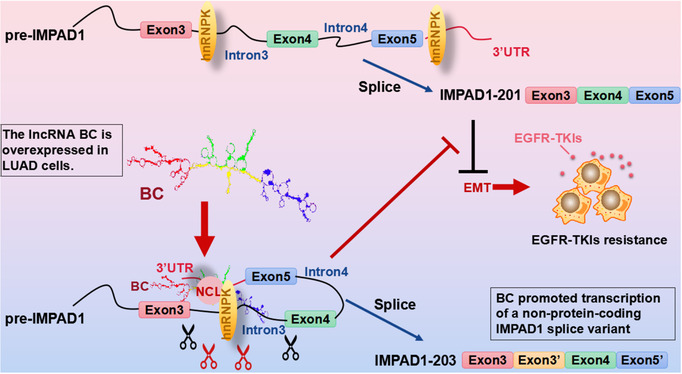
Results: In this study, we show that the long non-coding RNA BC009639 (BC) is involved in acquired resistance to EGFR-targeted therapies. Among the 235 long non-coding RNAs that were differentially expressed in lung cancer cell lines, with different metastatic potentials, BC promoted growth, invasion, metastasis, and resistance to EGFR-tyrosine kinase inhibitors (EGFR-TKIs), both in vitro and in vivo. BC was highly expressed in 428 patients with lung adenocarcinoma (LUAD) and high BC expression correlated with reduced efficacy of EGFR-TKI therapy. To uncover the molecular mechanism of BC-mediated EGFR-TKI resistance in lung cancer, we screened and identified nucleolin and hnRNPK that interact with BC. BC formed the splicing complex with nucleolin and hnRNPK to facilitate the production of a non-protein-coding inositol monophosphatase domain containing 1 (IMPAD1) splice variant, instead of the protein-coding variant. The BC-mediated alternative splicing (AS) of IMPAD1 resulted in the induction of the epithelial-mesenchymal transition and resistance to EGFR-TKI in lung cancer. High BC expression correlated with clinical progress and poor survival among 402 patients with LUAD.
Disscussion: Through alternative splicing, BC boosted the non-coding IMPAD1-203 transcript variant while suppressing the IMPAD1-201 variant. In order to control the processing of pre-mRNA, BC not only attracted RNA binding proteins (NCL, IGF2BP1) or splicing factors (hnRNPK), but also controlled the formation of the splicing-regulator complex by creating RNA-RNA-duplexes.
Conclusion: Our results reveal an important role for BC in mediating resistance to EGFR-targeted therapy in LUAD through IMPAD1 AS and in implication for the targeted therapy resistance.
Keywords: EGFR-TKI resistance; alternative splicing; inositol monophosphatase domain containing 1; lung adenocarcinoma; non-coding RNA.
© 2023 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors have declared that no conflicts of interest and no competing interest exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous