

The structure of DarB in complex with RelNTD reveals nonribosomal activation of Rel stringent factors
- PMID: 36652515
- PMCID: PMC9848473
- DOI: 10.1126/sciadv.ade4077
The structure of DarB in complex with RelNTD reveals nonribosomal activation of Rel stringent factors
Abstract
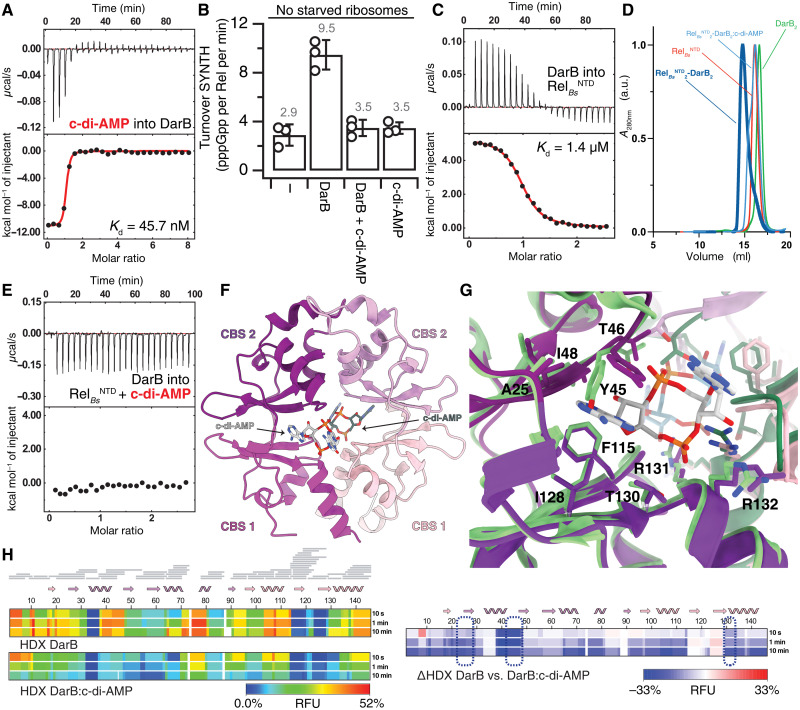
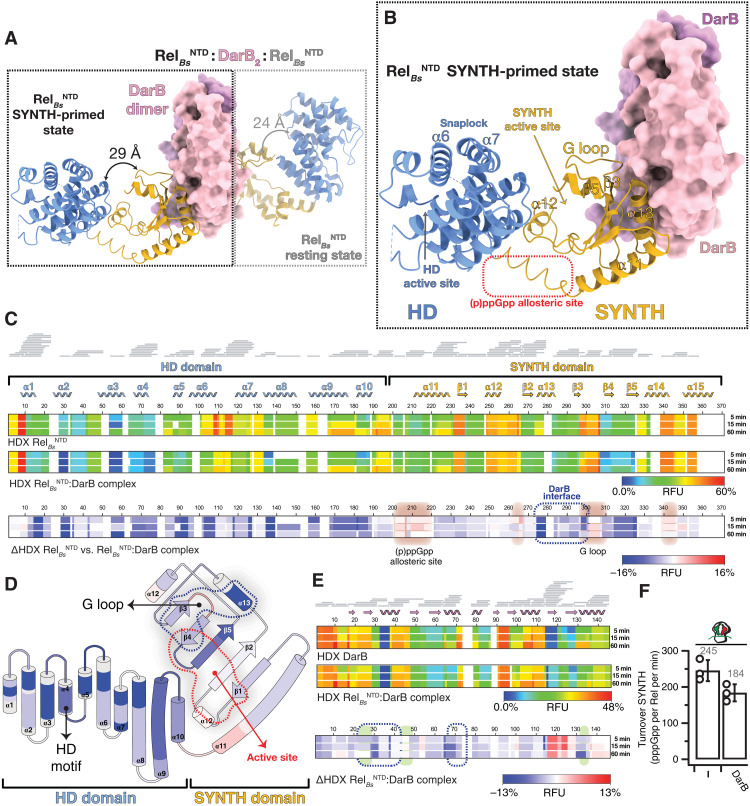
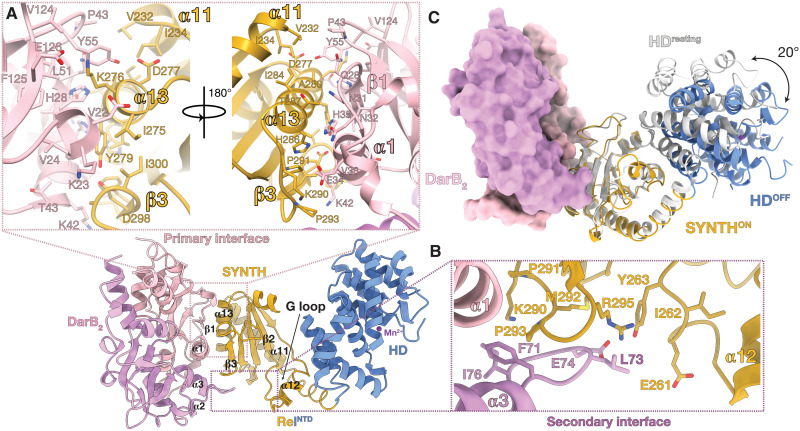
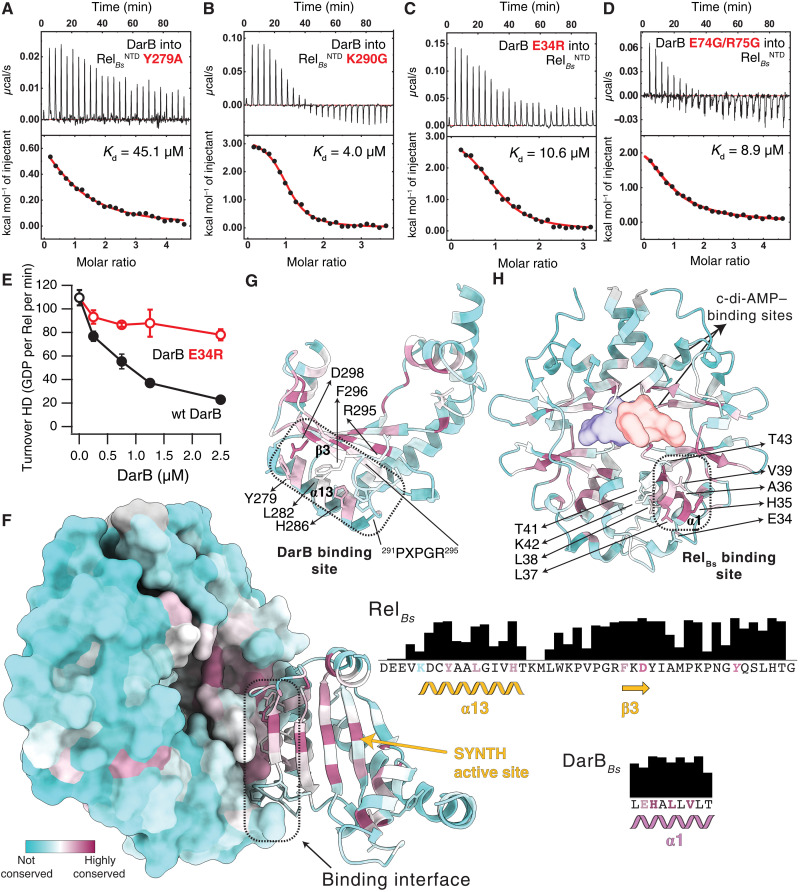
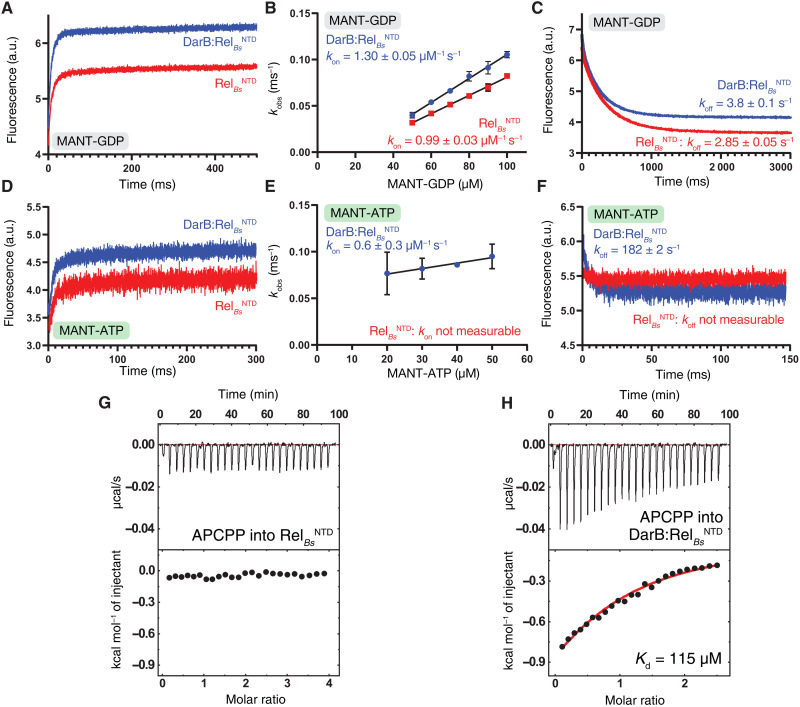
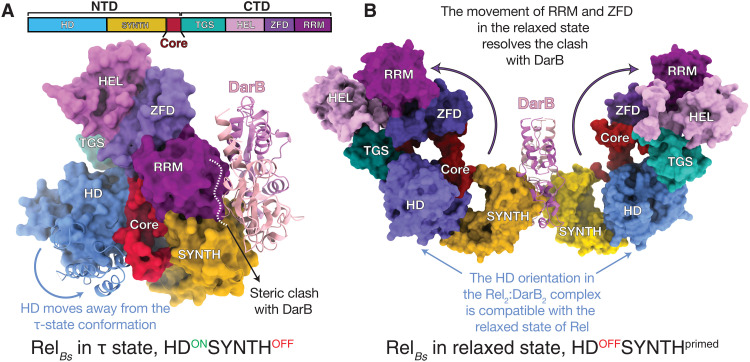
Rel stringent factors are bifunctional ribosome-associated enzymes that catalyze both synthesis and hydrolysis of the alarmones (p)ppGpp. Besides the allosteric control by starved ribosomes and (p)ppGpp, Rel is regulated by various protein factors depending on specific stress conditions, including the c-di-AMP-binding protein DarB. However, how these effector proteins control Rel remains unknown. We have determined the crystal structure of the DarB2:RelNTD2 complex, uncovering that DarB directly engages the SYNTH domain of Rel to stimulate (p)ppGpp synthesis. This association with DarB promotes a SYNTH-primed conformation of the N-terminal domain region, markedly increasing the affinity of Rel for ATP while switching off the hydrolase activity of the enzyme. Binding to c-di-AMP rigidifies DarB, imposing an entropic penalty that precludes DarB-mediated control of Rel during normal growth. Our experiments provide the basis for understanding a previously unknown mechanism of allosteric regulation of Rel stringent factors independent of amino acid starvation.
Figures
References
-
- S. E. Irving, N. R. Choudhury, R. M. Corrigan, The stringent response and physiological roles of (pp)pGpp in bacteria. Nat. Rev. Microbiol. 19, 256–271 (2021). - PubMed
-
- G. Bange, D. E. Brodersen, A. Liuzzi, W. Steinchen, Two P or not two P: Understanding regulation by the bacterial second messengers (p)ppGpp. Annu. Rev. Microbiol. 75, 383–406 (2021). - PubMed
-
- B. W. Anderson, D. K. Fung, J. D. Wang, Regulatory themes and variations by the stress-signaling nucleotide alarmones (p)ppGpp in bacteria. Annu. Rev. Genet. 55, 115–133 (2021). - PubMed
-
- G. Mittenhuber, Comparative genomics and evolution of genes encoding bacterial (p)ppGpp synthetases/hydrolases (the Rel, RelA and SpoT proteins). J. Mol. Microbiol. Biotechnol. 3, 585–600 (2001). - PubMed
